

Tissue-specific cell-free DNA degradation quantifies circulating tumor DNA burden
- PMID: 33850132
- PMCID: PMC8044092
- DOI: 10.1038/s41467-021-22463-y
Tissue-specific cell-free DNA degradation quantifies circulating tumor DNA burden
Abstract
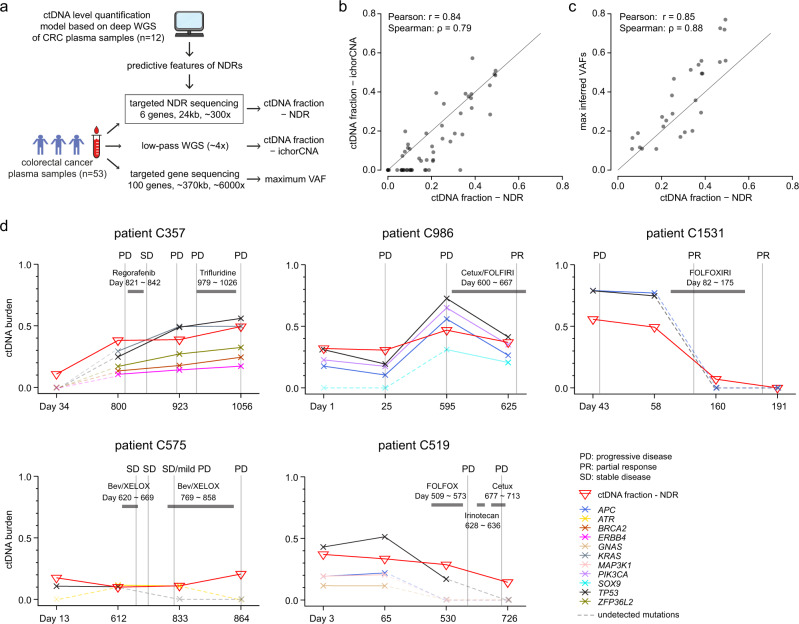
Profiling of circulating tumor DNA (ctDNA) may offer a non-invasive approach to monitor disease progression. Here, we develop a quantitative method, exploiting local tissue-specific cell-free DNA (cfDNA) degradation patterns, that accurately estimates ctDNA burden independent of genomic aberrations. Nucleosome-dependent cfDNA degradation at promoters and first exon-intron junctions is strongly associated with differential transcriptional activity in tumors and blood. A quantitative model, based on just 6 regulatory regions, could accurately predict ctDNA levels in colorectal cancer patients. Strikingly, a model restricted to blood-specific regulatory regions could predict ctDNA levels across both colorectal and breast cancer patients. Using compact targeted sequencing (<25 kb) of predictive regions, we demonstrate how the approach could enable quantitative low-cost tracking of ctDNA dynamics and disease progression.
Conflict of interest statement
The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
