

VirION2: a short- and long-read sequencing and informatics workflow to study the genomic diversity of viruses in nature
- PMID: 33850654
- PMCID: PMC8018248
- DOI: 10.7717/peerj.11088
VirION2: a short- and long-read sequencing and informatics workflow to study the genomic diversity of viruses in nature
Abstract
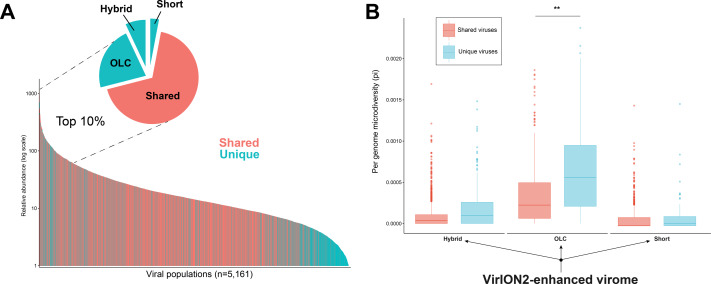
Microbes play fundamental roles in shaping natural ecosystem properties and functions, but do so under constraints imposed by their viral predators. However, studying viruses in nature can be challenging due to low biomass and the lack of universal gene markers. Though metagenomic short-read sequencing has greatly improved our virus ecology toolkit-and revealed many critical ecosystem roles for viruses-microdiverse populations and fine-scale genomic traits are missed. Some of these microdiverse populations are abundant and the missed regions may be of interest for identifying selection pressures that underpin evolutionary constraints associated with hosts and environments. Though long-read sequencing promises complete virus genomes on single reads, it currently suffers from high DNA requirements and sequencing errors that limit accurate gene prediction. Here we introduce VirION2, an integrated short- and long-read metagenomic wet-lab and informatics pipeline that updates our previous method (VirION) to further enhance the utility of long-read viral metagenomics. Using a viral mock community, we first optimized laboratory protocols (polymerase choice, DNA shearing size, PCR cycling) to enable 76% longer reads (now median length of 6,965 bp) from 100-fold less input DNA (now 1 nanogram). Using a virome from a natural seawater sample, we compared viromes generated with VirION2 against other library preparation options (unamplified, original VirION, and short-read), and optimized downstream informatics for improved long-read error correction and assembly. VirION2 assemblies combined with short-read based data ('enhanced' viromes), provided significant improvements over VirION libraries in the recovery of longer and more complete viral genomes, and our optimized error-correction strategy using long- and short-read data achieved 99.97% accuracy. In the seawater virome, VirION2 assemblies captured 5,161 viral populations (including all of the virus populations observed in the other assemblies), 30% of which were uniquely assembled through inclusion of long-reads, and 22% of the top 10% most abundant virus populations derived from assembly of long-reads. Viral populations unique to VirION2 assemblies had significantly higher microdiversity means, which may explain why short-read virome approaches failed to capture them. These findings suggest the VirION2 sample prep and workflow can help researchers better investigate the virosphere, even from challenging low-biomass samples. Our new protocols are available to the research community on protocols.io as a 'living document' to facilitate dissemination of updates to keep pace with the rapid evolution of long-read sequencing technology.
Keywords: Long-reads; Metagenome; Nanopore sequencing; Phage; Viral metagenomics; Virome; Virus.
©2021 Zablocki et al.
Conflict of interest statement
The authors declare there are no competing interests.
Figures




Similar articles
-
Long-read viral metagenomics captures abundant and microdiverse viral populations and their niche-defining genomic islands.PeerJ. 2019 Apr 25;7:e6800. doi: 10.7717/peerj.6800. eCollection 2019. PeerJ. 2019. PMID: 31086738 Free PMC article.
-
Long-Read Metagenomics Improves the Recovery of Viral Diversity from Complex Natural Marine Samples.mSystems. 2022 Jun 28;7(3):e0019222. doi: 10.1128/msystems.00192-22. Epub 2022 Jun 13. mSystems. 2022. PMID: 35695508 Free PMC article.
-
The long and short of it: benchmarking viromics using Illumina, Nanopore and PacBio sequencing technologies.Microb Genom. 2024 Feb;10(2):001198. doi: 10.1099/mgen.0.001198. Microb Genom. 2024. PMID: 38376377 Free PMC article.
-
Emerging technologies in the study of the virome.Curr Opin Virol. 2022 Jun;54:101231. doi: 10.1016/j.coviro.2022.101231. Epub 2022 May 25. Curr Opin Virol. 2022. PMID: 35643020 Review.
-
Evolution of selective-sequencing approaches for virus discovery and virome analysis.Virus Res. 2017 Jul 15;239:172-179. doi: 10.1016/j.virusres.2017.06.005. Epub 2017 Jun 3. Virus Res. 2017. PMID: 28583442 Free PMC article. Review.
Cited by
-
Long-read powered viral metagenomics in the oligotrophic Sargasso Sea.Nat Commun. 2024 May 14;15(1):4089. doi: 10.1038/s41467-024-48300-6. Nat Commun. 2024. PMID: 38744831 Free PMC article.
-
Evolutionary Divergence of Marinobacter Strains in Cryopeg Brines as Revealed by Pangenomics.Front Microbiol. 2022 Jun 6;13:879116. doi: 10.3389/fmicb.2022.879116. eCollection 2022. Front Microbiol. 2022. PMID: 35733954 Free PMC article.
-
Databases, Knowledgebases, and Software Tools for Virus Informatics.Adv Exp Med Biol. 2022;1368:1-19. doi: 10.1007/978-981-16-8969-7_1. Adv Exp Med Biol. 2022. PMID: 35594018
-
Lower viral evolutionary pressure under stable versus fluctuating conditions in subzero Arctic brines.Microbiome. 2023 Aug 7;11(1):174. doi: 10.1186/s40168-023-01619-6. Microbiome. 2023. PMID: 37550784 Free PMC article.
-
Simple, reference-independent assessment to empirically guide correction and polishing of hybrid microbial community metagenomic assembly.PeerJ. 2024 Nov 8;12:e18132. doi: 10.7717/peerj.18132. eCollection 2024. PeerJ. 2024. PMID: 39529629 Free PMC article.
References
-
- Al-Shayeb B, Sachdeva R, Chen L-X, Ward F, Munk P, Devoto A, Castelle CJ, Olm MR, Bouma-Gregson K, Amano Y, He C, Méheust R, Brooks B, Thomas A, Lavy A, Matheus-Carnevali P, Sun C, Goltsman DSA, Borton MA, Sharrar A, Jaffe AL, Nelson TC, Kantor R, Keren R, Lane KR, Farag IF, Lei S, Finstad K, Amundson R, Anantharaman K, Zhou J, Probst AJ, Power ME, Tringe SG, Li W-J, Wrighton K, Harrison S, Morowitz M, Relman DA, Doudna JA, Lehours A-C, Warren L, Cate JHD, Santini JM, Banfield JF. Clades of huge phages from across Earth’s ecosystems. Nature. 2020;578:425–431. doi: 10.1038/s41586-020-2007-4. - DOI - PMC - PubMed
-
- Beaulaurier J, Luo E, Eppley JM, Den UylP, Dai X, Burger A, Turner DJ, Pendelton M, Juul S, Harrington E, DeLong EF. Assembly-free single-molecule sequencing recovers complete virus genomes from natural microbial communities. Genome Research. 2020;30:437–446. doi: 10.1101/gr.251686.119. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
