

This is a preprint.
Antibodies to the SARS-CoV-2 receptor-binding domain that maximize breadth and resistance to viral escape
- PMID: 33851154
- PMCID: PMC8043444
- DOI: 10.1101/2021.04.06.438709
Antibodies to the SARS-CoV-2 receptor-binding domain that maximize breadth and resistance to viral escape
Update in
-
SARS-CoV-2 RBD antibodies that maximize breadth and resistance to escape.Nature. 2021 Sep;597(7874):97-102. doi: 10.1038/s41586-021-03807-6. Epub 2021 Jul 14. Nature. 2021. PMID: 34261126 Free PMC article.
Abstract
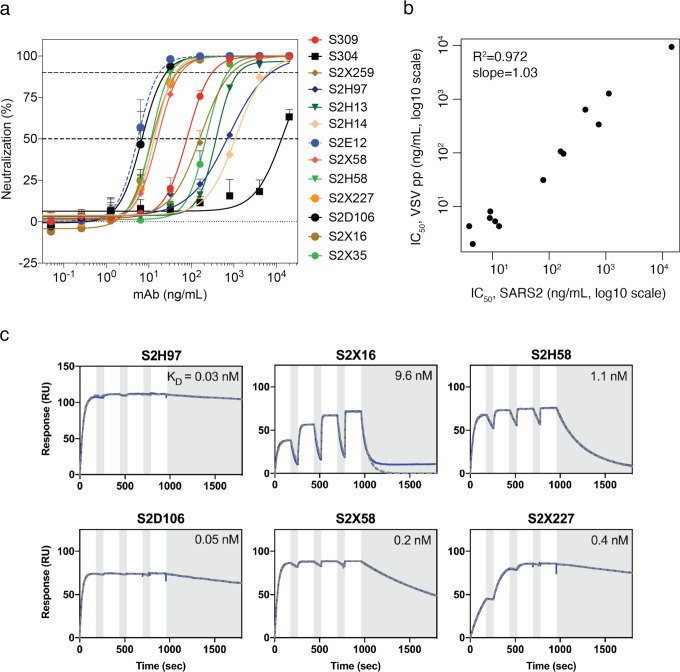
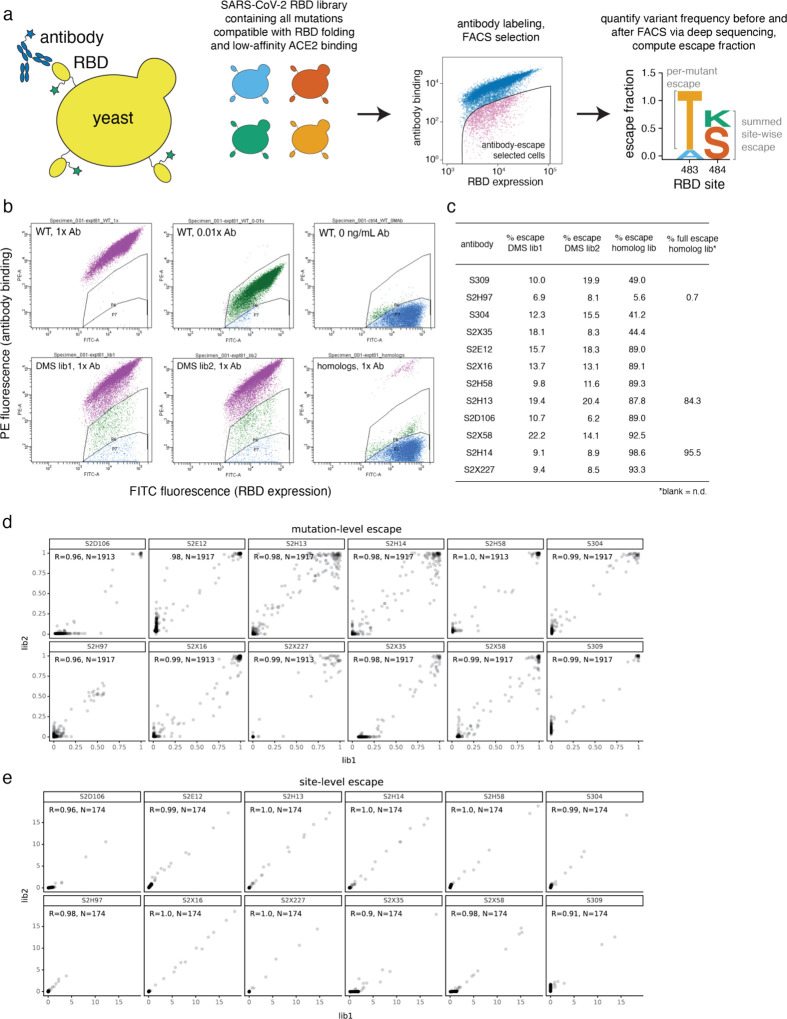
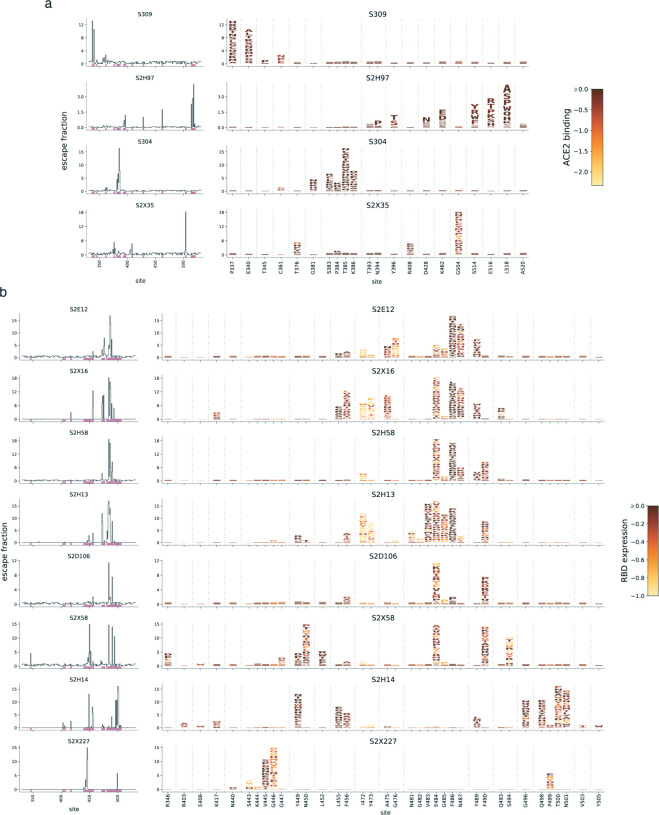
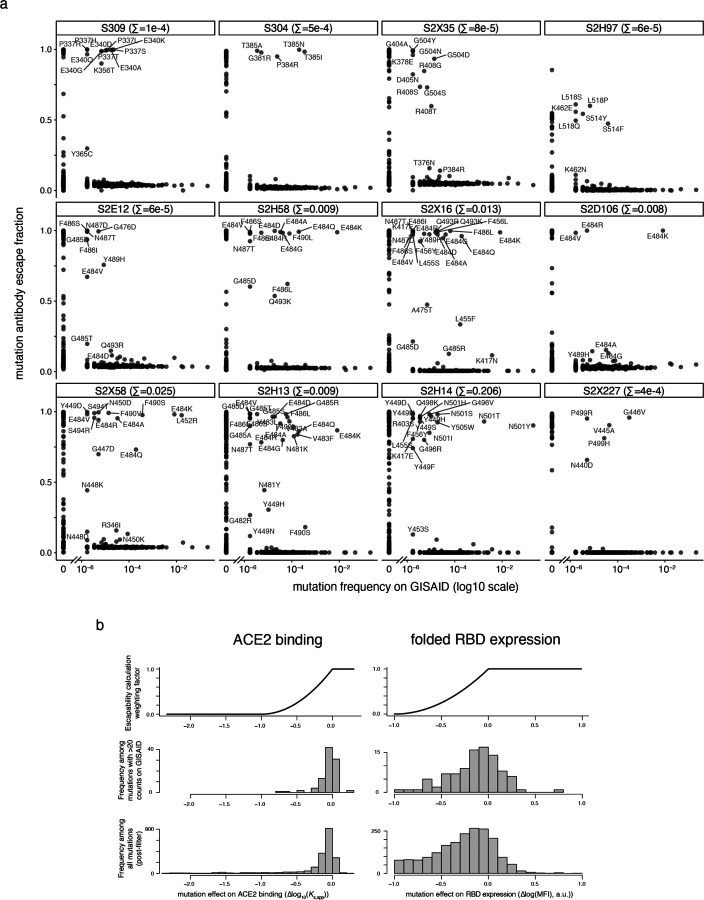
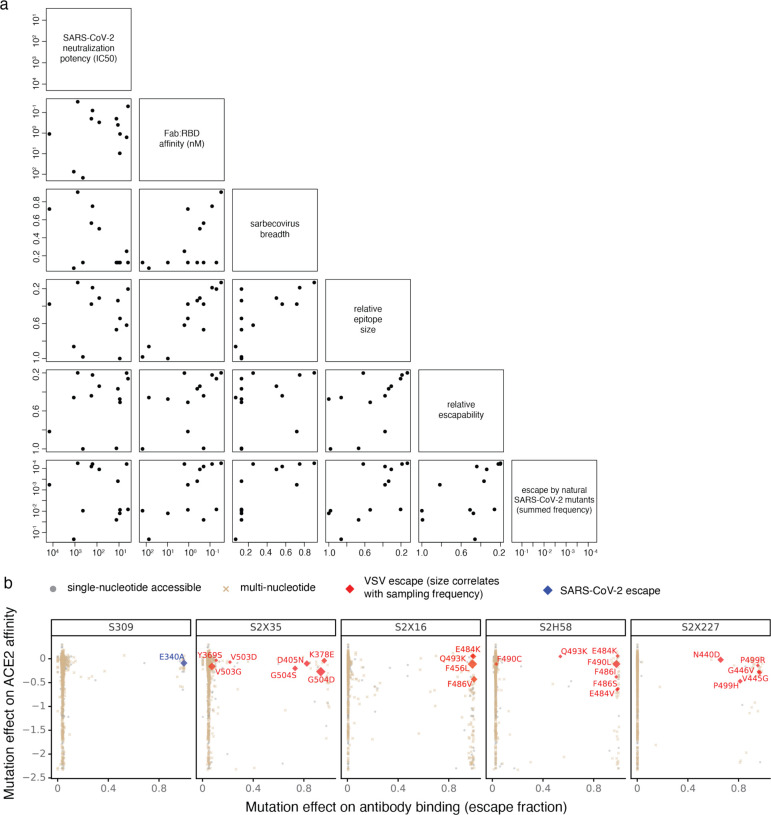
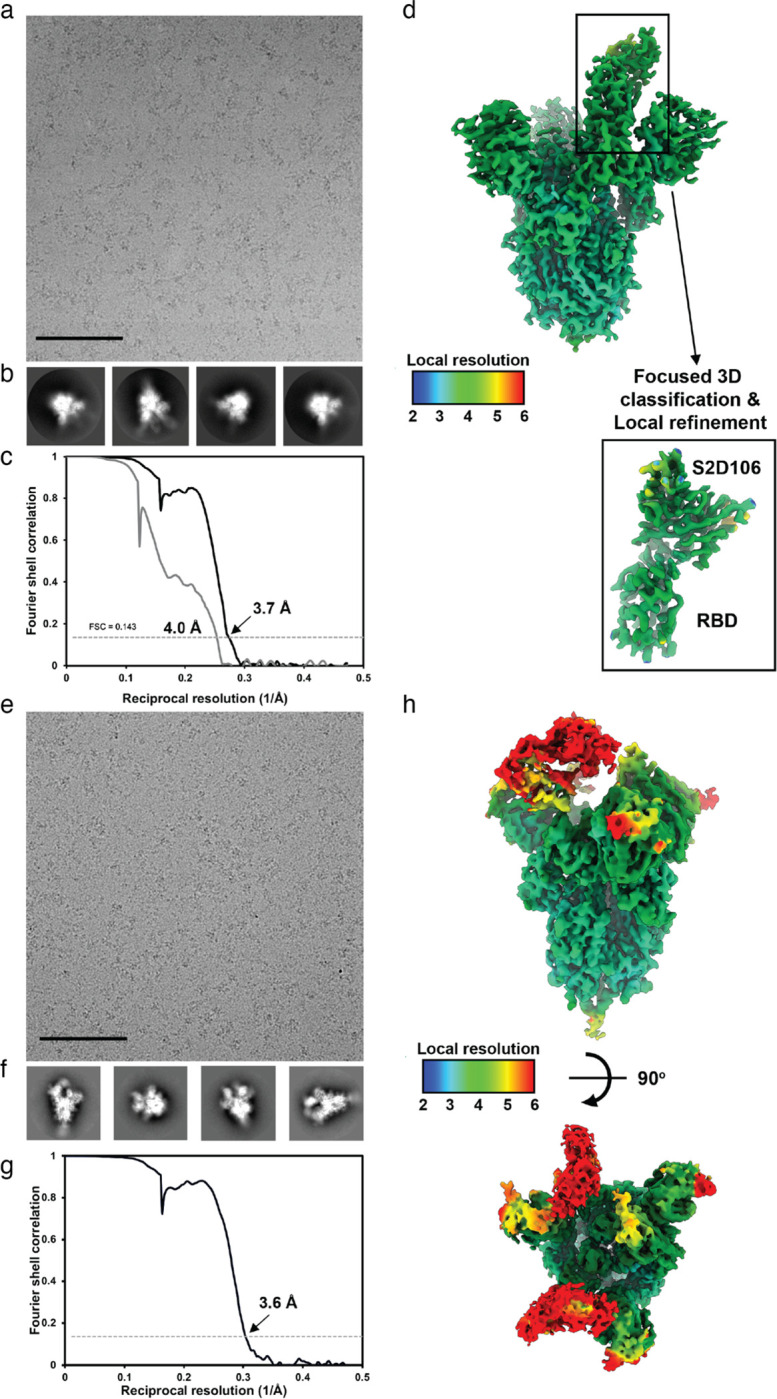
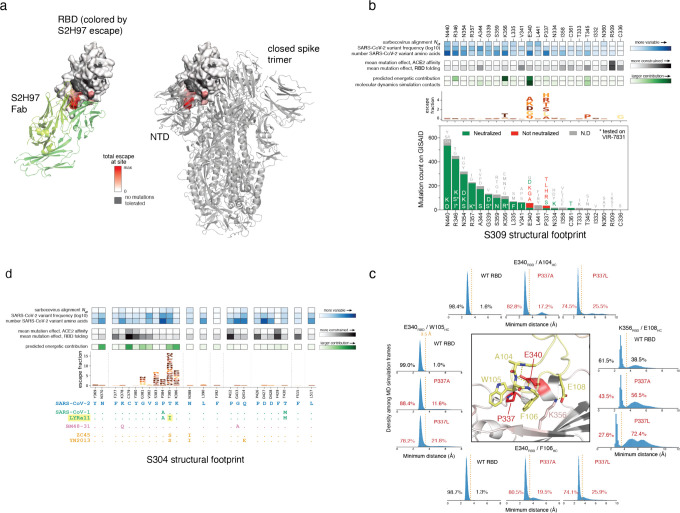
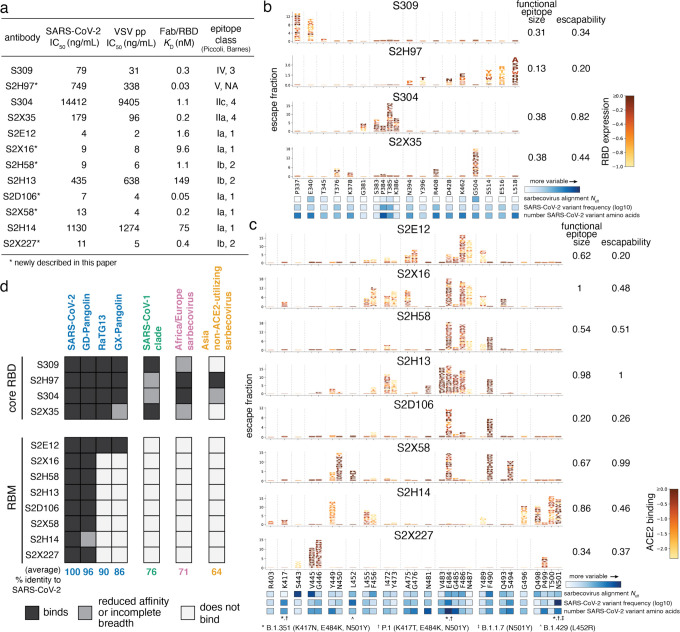
An ideal anti-SARS-CoV-2 antibody would resist viral escape 1-3 , have activity against diverse SARS-related coronaviruses 4-7 , and be highly protective through viral neutralization 8-11 and effector functions 12,13 . Understanding how these properties relate to each other and vary across epitopes would aid development of antibody therapeutics and guide vaccine design. Here, we comprehensively characterize escape, breadth, and potency across a panel of SARS-CoV-2 antibodies targeting the receptor-binding domain (RBD), including S309 4 , the parental antibody of the late-stage clinical antibody VIR-7831. We observe a tradeoff between SARS-CoV-2 in vitro neutralization potency and breadth of binding across SARS-related coronaviruses. Nevertheless, we identify several neutralizing antibodies with exceptional breadth and resistance to escape, including a new antibody (S2H97) that binds with high affinity to all SARS-related coronavirus clades via a unique RBD epitope centered on residue E516. S2H97 and other escape-resistant antibodies have high binding affinity and target functionally constrained RBD residues. We find that antibodies targeting the ACE2 receptor binding motif (RBM) typically have poor breadth and are readily escaped by mutations despite high neutralization potency, but we identify one potent RBM antibody (S2E12) with breadth across sarbecoviruses closely related to SARS-CoV-2 and with a high barrier to viral escape. These data highlight functional diversity among antibodies targeting the RBD and identify epitopes and features to prioritize for antibody and vaccine development against the current and potential future pandemics.
Figures

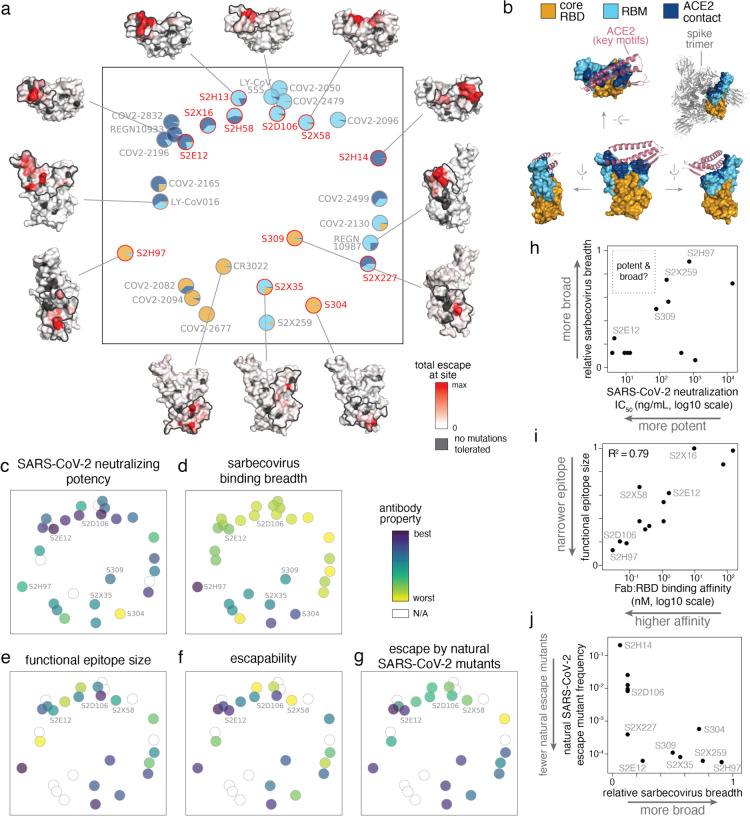
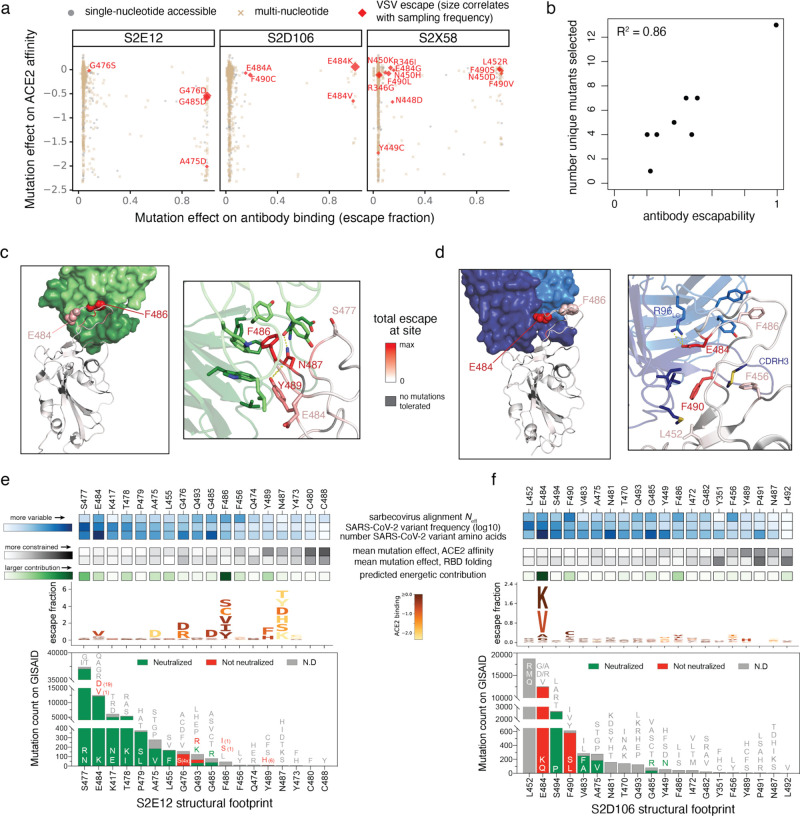
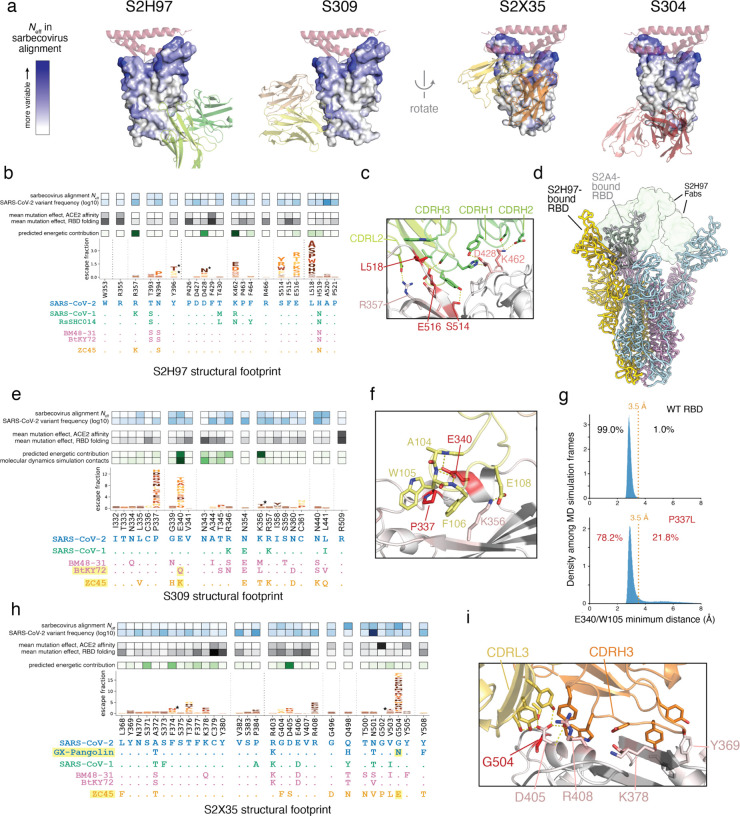
References
-
- Pinto D. et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 583, 290–295 (2020). - PubMed
Publication types
Grants and funding
- R01 GM120553/GM/NIGMS NIH HHS/United States
- DP1 AI158186/AI/NIAID NIH HHS/United States
- P30 GM124169/GM/NIGMS NIH HHS/United States
- S10 OD028685/OD/NIH HHS/United States
- P30 GM133894/GM/NIGMS NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- R01 AI127893/AI/NIAID NIH HHS/United States
- T32 AI083203/AI/NIAID NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- R01 AI141707/AI/NIAID NIH HHS/United States
- R01 GM121505/GM/NIGMS NIH HHS/United States
- R01 GM132386/GM/NIGMS NIH HHS/United States
- HHSN272201700059C/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous