

PINK1 mediates the protective effects of thyroid hormone T3 in hyperoxia-induced lung injury
- PMID: 33851544
- PMCID: PMC8285622
- DOI: 10.1152/ajplung.00598.2020
PINK1 mediates the protective effects of thyroid hormone T3 in hyperoxia-induced lung injury
Abstract
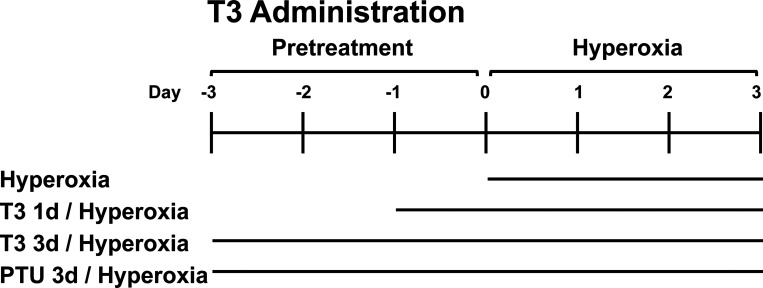
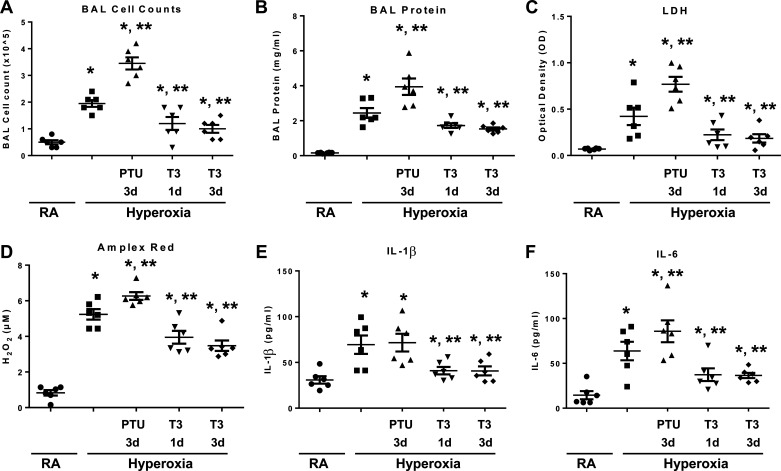
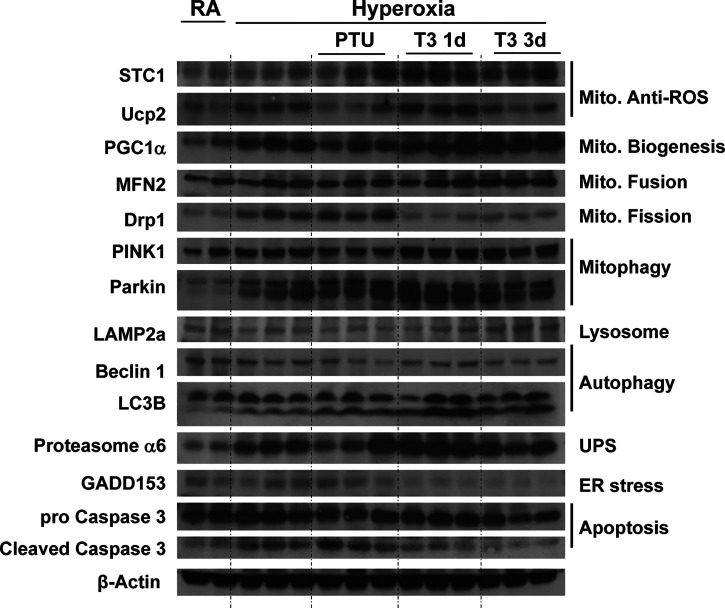
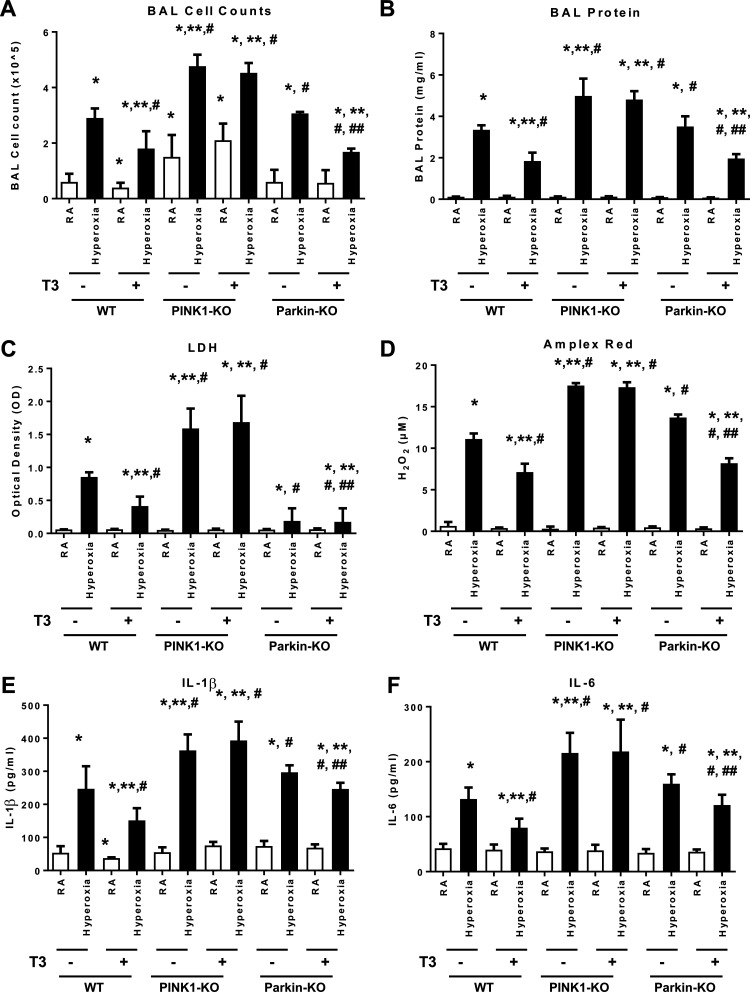
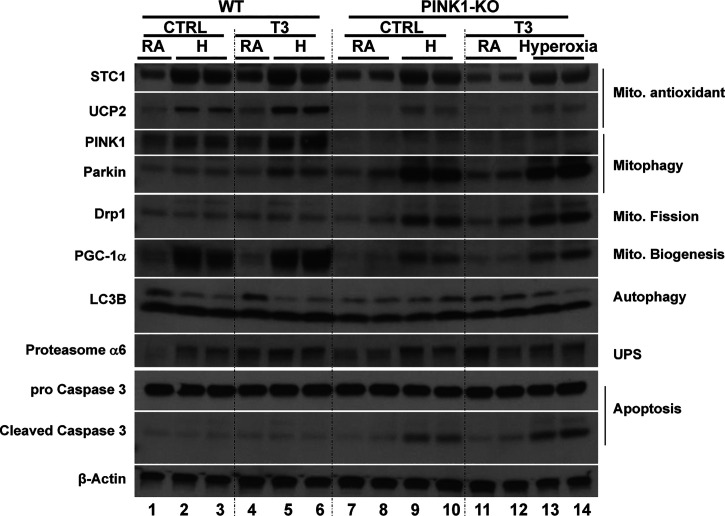
Hyperoxia can lead to respiratory failure and death. Our previous work demonstrates that oxidant and mitochondrial injury play a critical role in hyperoxia-induced acute lung injury (HALI). Recently, thyroid hormone has been demonstrated to promote mitochondrial survival in other models of lung injury, but its role in hyperoxia is unknown. Adult wild-type (WT) mice were pretreated with either nebulized triiodothyronine (T3, 40 μg/kg) for 1 or 3 days, or with propylthiouracil (PTU, 100 μg/kg), for 3 days. Following pretreatment, WT mice underwent 72 h of hyperoxia exposure. WT and PINK1-/- mice were pretreated with either nebulized T3 (40 μg/kg) for 3 days or no pretreatment before 72 h continuous hyperoxia exposure. Bronchoalveolar lavage (BAL), histological changes in cellular composition, and type I cytokine induction were assessed. Lung lysates for mitochondrial cellular bioenergetics markers were analyzed by Western blot. Hyperoxia caused a significant increase in BAL total cell counts and lung cellular infiltrates. Administration of PTU enhanced HALI, whereas T3 attenuated HALI, inflammation, and oxidants in WT mice. In addition, T3 pretreatment increased mitochondrial biogenesis/fusion/mitophagy and decreased ER stress and apoptosis. PINK1-/- mice were more susceptible to hyperoxia than WT mice. Notably, pretreatment with T3 did not attenuate HALI in PINK1-/- mice. In addition, T3 pretreatment increased mitochondrial anti-ROS potential, improved mitochondrial bioenergetics and mitophagy, and attenuated mitochondria-regulated apoptosis, all in a PINK1-dependent manner. Our results highlight a novel protective role for PINK1 in mediating the cytoprotective effects of thyroid hormone in HALI. Therefore, thyroid hormone may represent a potential therapy for ALI.
Keywords: HALI; PINK1; hyperoxia; lung injury; thyroid hormone.
Conflict of interest statement
Y.Z., G.Y., N.K., and P.J.L. have IP on the use of thyroid hormone mimetics in IPF and ARDS licensed to biotechnology. There are no conflicts of interest, financial or otherwise, to disclose.
Figures

References
-
- Yau WW, Singh BK, Lesmana R, Zhou J, Sinha RA, Wong KA, Wu Y, Bay B-H, Sugii S, Sun L, Yen PM. Thyroid hormone (T3) stimulates brown adipose tissue activation via mitochondrial biogenesis and MTOR-mediated mitophagy. Autophagy 15: 131–150, 2019. doi: 10.1080/15548627.2018.1511263. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases