

Reducing acetylated tau is neuroprotective in brain injury
- PMID: 33852912
- PMCID: PMC8491234
- DOI: 10.1016/j.cell.2021.03.032
Reducing acetylated tau is neuroprotective in brain injury
Abstract
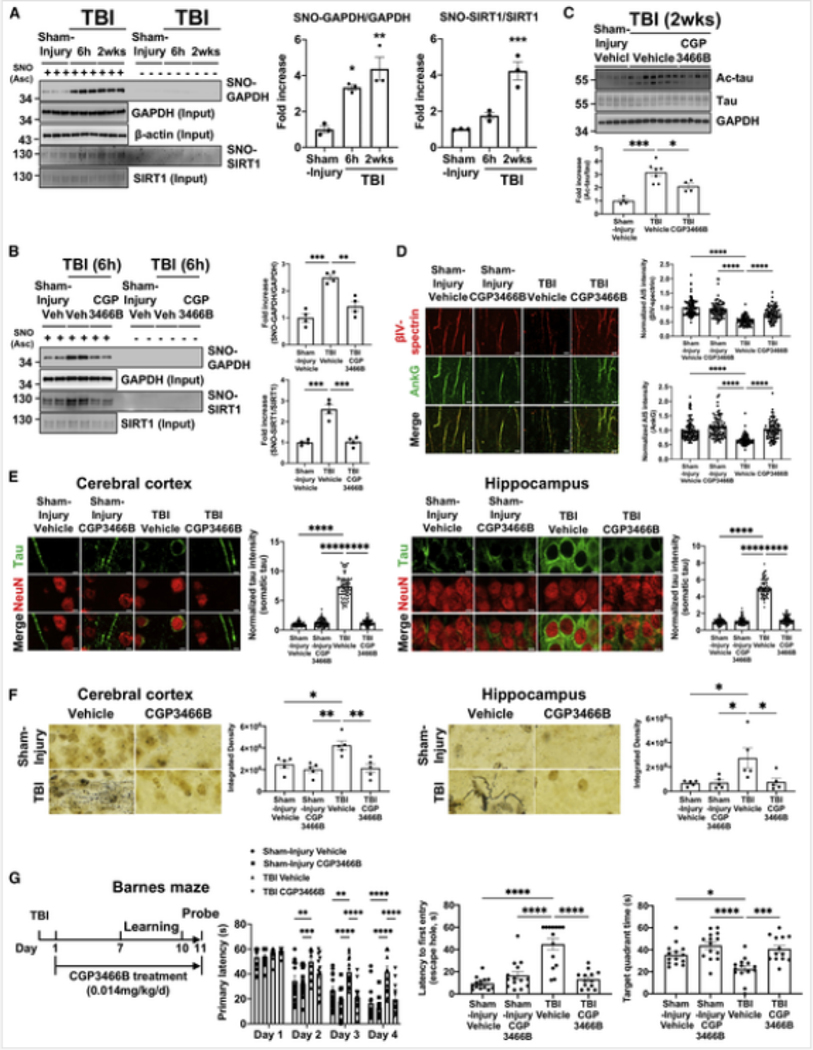
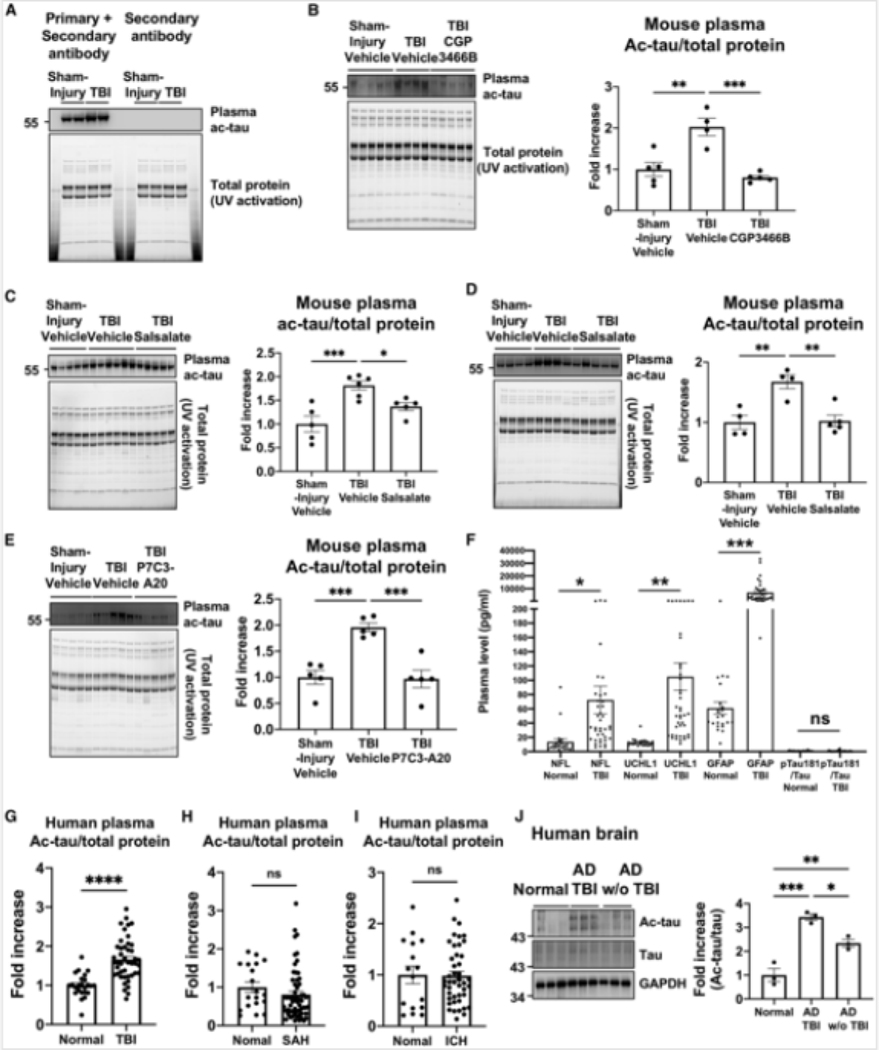
Traumatic brain injury (TBI) is the largest non-genetic, non-aging related risk factor for Alzheimer's disease (AD). We report here that TBI induces tau acetylation (ac-tau) at sites acetylated also in human AD brain. This is mediated by S-nitrosylated-GAPDH, which simultaneously inactivates Sirtuin1 deacetylase and activates p300/CBP acetyltransferase, increasing neuronal ac-tau. Subsequent tau mislocalization causes neurodegeneration and neurobehavioral impairment, and ac-tau accumulates in the blood. Blocking GAPDH S-nitrosylation, inhibiting p300/CBP, or stimulating Sirtuin1 all protect mice from neurodegeneration, neurobehavioral impairment, and blood and brain accumulation of ac-tau after TBI. Ac-tau is thus a therapeutic target and potential blood biomarker of TBI that may represent pathologic convergence between TBI and AD. Increased ac-tau in human AD brain is further augmented in AD patients with history of TBI, and patients receiving the p300/CBP inhibitors salsalate or diflunisal exhibit decreased incidence of AD and clinically diagnosed TBI.
Keywords: Alzheimer’s disease; P7C3; acetylation; congenital muscular dystrophy; diflunisal; neurodegeneration; neuroprotection; omigapil; salsalate; tau; traumatic brain injury.
Published by Elsevier Inc.
Conflict of interest statement
Declaration of interests A.A.P. is an inventor on patents related to P7C3. L.G. is a founder of Aeton Therapeutics. No other authors declare competing interests.
Figures





Comment in
-
Targeting tau in traumatic brain injury.Nat Rev Drug Discov. 2021 Jun;20(6):424. doi: 10.1038/d41573-021-00070-2. Nat Rev Drug Discov. 2021. PMID: 33888906 No abstract available.
-
A shared pathway?Nat Rev Neurosci. 2021 Jun;22(6):325. doi: 10.1038/s41583-021-00471-7. Nat Rev Neurosci. 2021. PMID: 33953376 No abstract available.
-
Acetylated tau: A missing link between head injury and dementia.Med. 2021 Jun 11;2(6):637-639. doi: 10.1016/j.medj.2021.05.005. Epub 2021 Jun 15. Med. 2021. PMID: 35590136
References
-
- Arun P, Abu-Taleb R, Oguntayo S, Tanaka M, Wang Y, Valiyaveettil M, Long JB, Zhang Y, Nambiar MP, 2013. Distinct patterns of expression of traumatic brain injury biomarkers after blast exposure: role of compromised cell membrane integrity. Neurosci. Lett. 552, 87–91. - PubMed
-
- Centers for Disease Control and Prevention, 2015. Report to Congress on Traumatic Brain Injury in the United States: Epidemiology and Rehabilitation. National Center for Injury Prevention and Control: Division of Unintentional Injury Prevention.
Publication types
MeSH terms
Substances
Grants and funding
- P30 AG013854/AG/NIA NIH HHS/United States
- F32 AG043301/AG/NIA NIH HHS/United States
- UL1 TR001412/TR/NCATS NIH HHS/United States
- P01 CA217797/CA/NCI NIH HHS/United States
- IK2 RX002928/RX/RRD VA/United States
- K01 AG057862/AG/NIA NIH HHS/United States
- I01 BX002439/BX/BLRD VA/United States
- R01 AG062566/AG/NIA NIH HHS/United States
- R01 NS098740/NS/NINDS NIH HHS/United States
- T32 GM007250/GM/NIGMS NIH HHS/United States
- K08 AG065463/AG/NIA NIH HHS/United States
- P30 AG062428/AG/NIA NIH HHS/United States
- R01 AG066707/AG/NIA NIH HHS/United States
- T32 CA078586/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
