

Spine impairment in mice high-expressing neuregulin 1 due to LIMK1 activation
- PMID: 33854034
- PMCID: PMC8047019
- DOI: 10.1038/s41419-021-03687-8
Spine impairment in mice high-expressing neuregulin 1 due to LIMK1 activation
Abstract
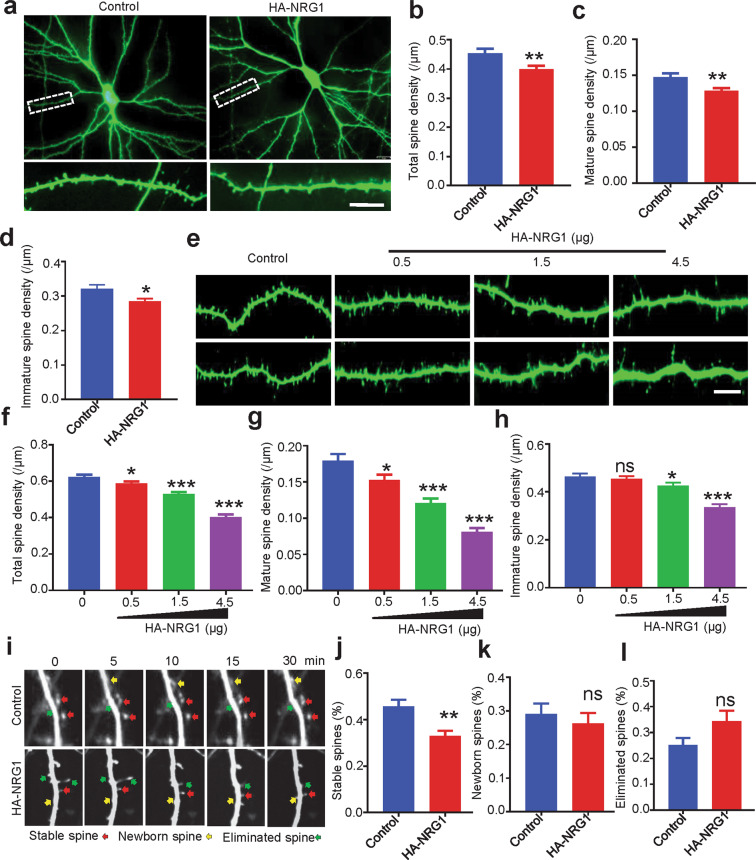
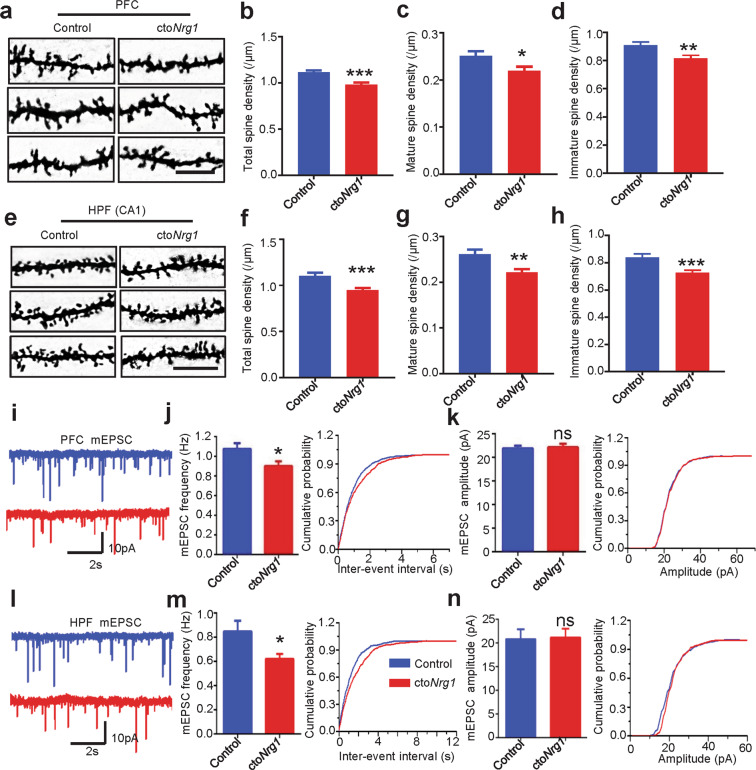
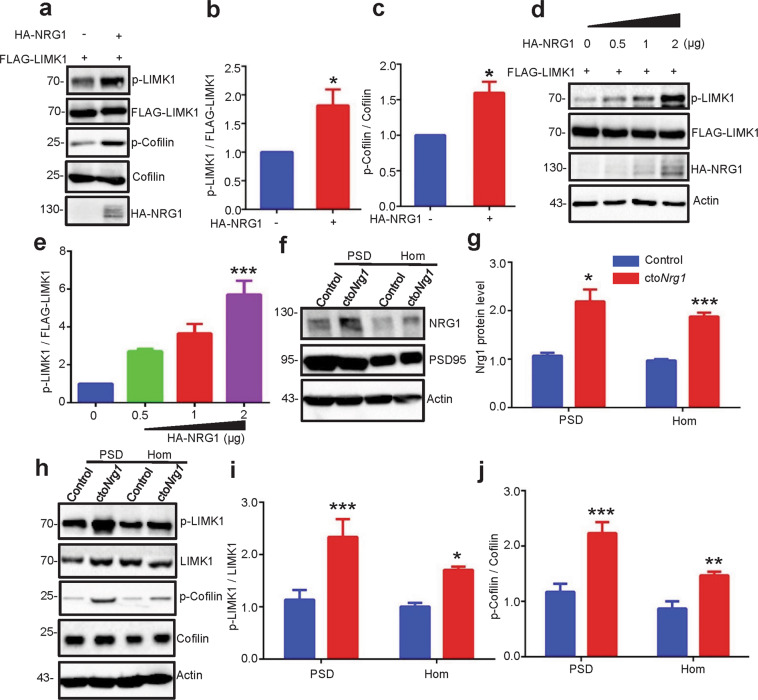
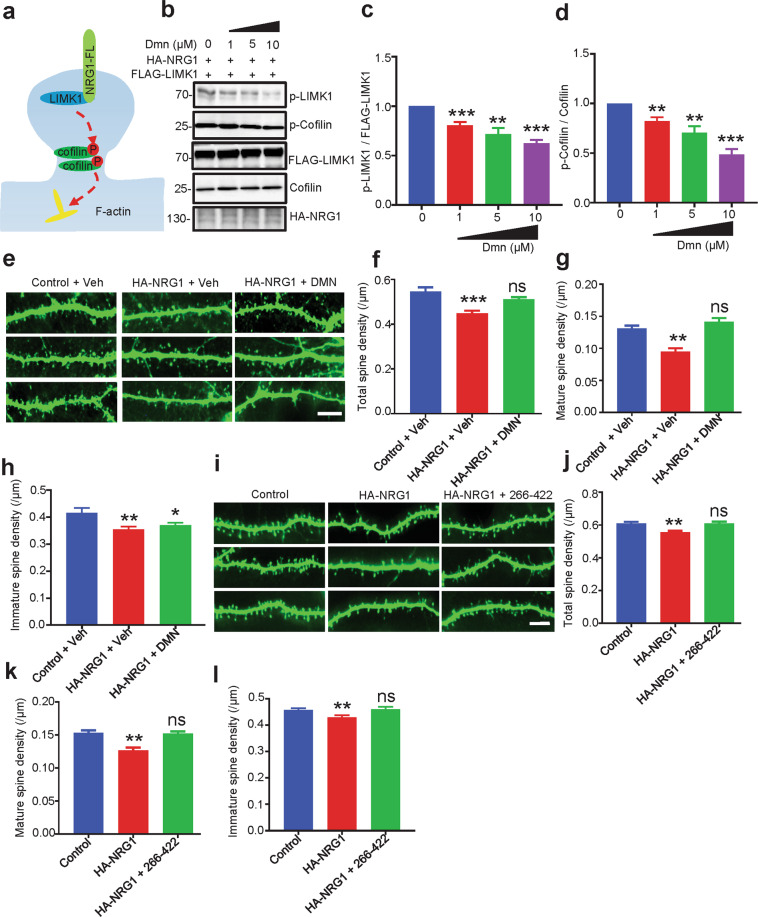
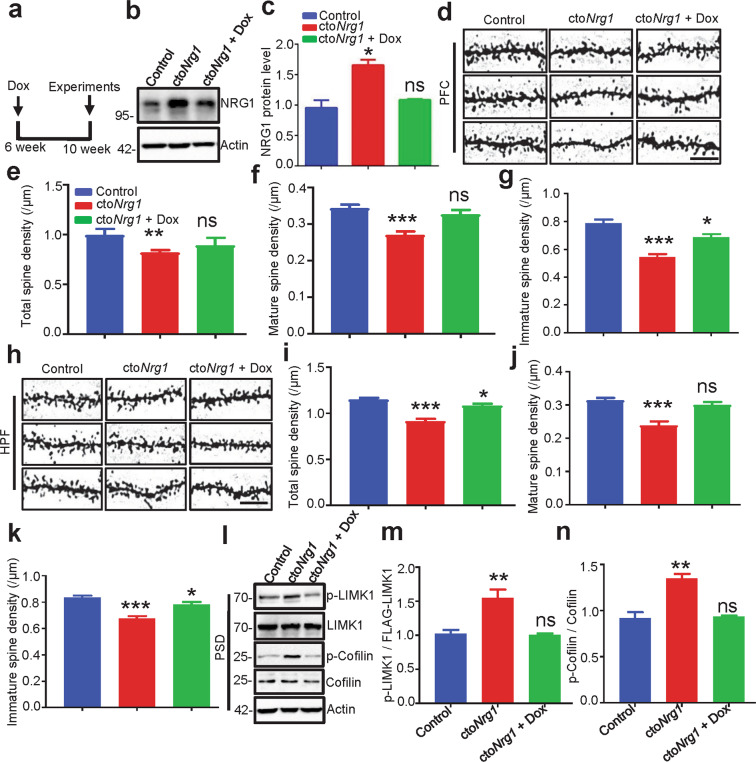
The genes encoding for neuregulin1 (NRG1), a growth factor, and its receptor ErbB4 are both risk factors of major depression disorder and schizophrenia (SZ). They have been implicated in neural development and synaptic plasticity. However, exactly how NRG1 variations lead to SZ remains unclear. Indeed, NRG1 levels are increased in postmortem brain tissues of patients with brain disorders. Here, we studied the effects of high-level NRG1 on dendritic spine development and function. We showed that spine density in the prefrontal cortex and hippocampus was reduced in mice (ctoNrg1) that overexpressed NRG1 in neurons. The frequency of miniature excitatory postsynaptic currents (mEPSCs) was reduced in both brain regions of ctoNrg1 mice. High expression of NRG1 activated LIMK1 and increased cofilin phosphorylation in postsynaptic densities. Spine reduction was attenuated by inhibiting LIMK1 or blocking the NRG1-LIMK1 interaction, or by restoring NRG1 protein level. These results indicate that a normal NRG1 protein level is necessary for spine homeostasis and suggest a pathophysiological mechanism of abnormal spines in relevant brain disorders.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
