

Consensus statement on standards and guidelines for the molecular diagnostics of Alport syndrome: refining the ACMG criteria
- PMID: 33854215
- PMCID: PMC8384871
- DOI: 10.1038/s41431-021-00858-1
Consensus statement on standards and guidelines for the molecular diagnostics of Alport syndrome: refining the ACMG criteria
Erratum in
-
Correction: Consensus statement on standards and guidelines for the molecular diagnostics of Alport syndrome: refining the ACMG criteria.Eur J Hum Genet. 2024 Jan;32(1):132. doi: 10.1038/s41431-023-01288-x. Eur J Hum Genet. 2024. PMID: 36721056 Free PMC article. No abstract available.
Abstract
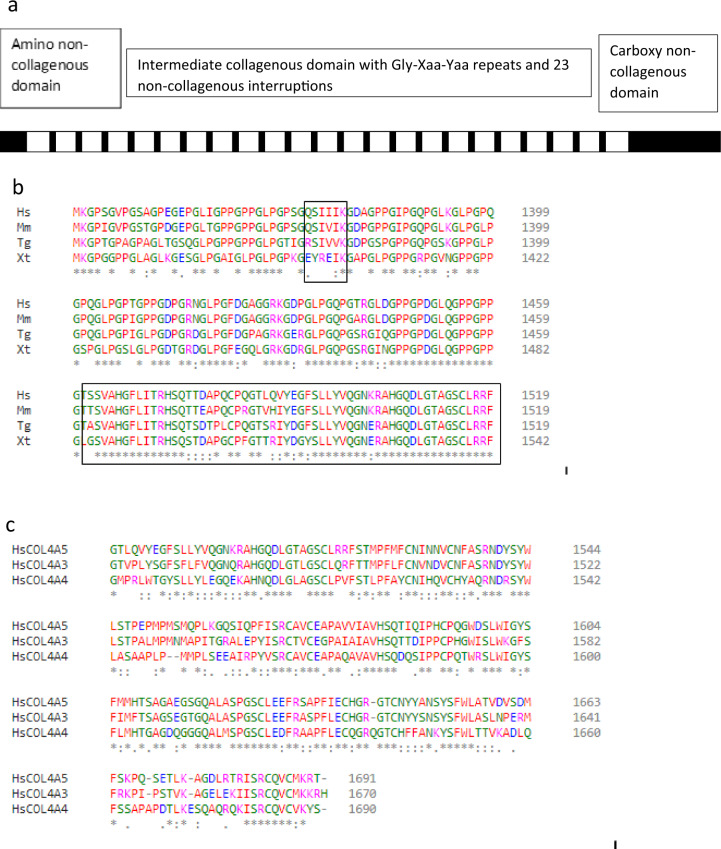
The recent Chandos House meeting of the Alport Variant Collaborative extended the indications for screening for pathogenic variants in the COL4A5, COL4A3 and COL4A4 genes beyond the classical Alport phenotype (haematuria, renal failure; family history of haematuria or renal failure) to include persistent proteinuria, steroid-resistant nephrotic syndrome, focal and segmental glomerulosclerosis (FSGS), familial IgA glomerulonephritis and end-stage kidney failure without an obvious cause. The meeting refined the ACMG criteria for variant assessment for the Alport genes (COL4A3-5). It identified 'mutational hotspots' (PM1) in the collagen IV α5, α3 and α4 chains including position 1 Glycine residues in the Gly-X-Y repeats in the intermediate collagenous domains; and Cysteine residues in the carboxy non-collagenous domain (PP3). It considered that 'well-established' functional assays (PS3, BS3) were still mainly research tools but sequencing and minigene assays were commonly used to confirm splicing variants. It was not possible to define the Minor Allele Frequency (MAF) threshold above which variants were considered Benign (BA1, BS1), because of the different modes of inheritances of Alport syndrome, and the occurrence of hypomorphic variants (often Glycine adjacent to a non-collagenous interruption) and local founder effects. Heterozygous COL4A3 and COL4A4 variants were common 'incidental' findings also present in normal reference databases. The recognition and interpretation of hypomorphic variants in the COL4A3-COL4A5 genes remains a challenge.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
