

Optimizing Bedaquiline for cardiotoxicity by structure based virtual screening, DFT analysis and molecular dynamic simulation studies to identify selective MDR-TB inhibitors
- PMID: 33854869
- PMCID: PMC7988025
- DOI: 10.1007/s40203-021-00086-x
Optimizing Bedaquiline for cardiotoxicity by structure based virtual screening, DFT analysis and molecular dynamic simulation studies to identify selective MDR-TB inhibitors
Abstract
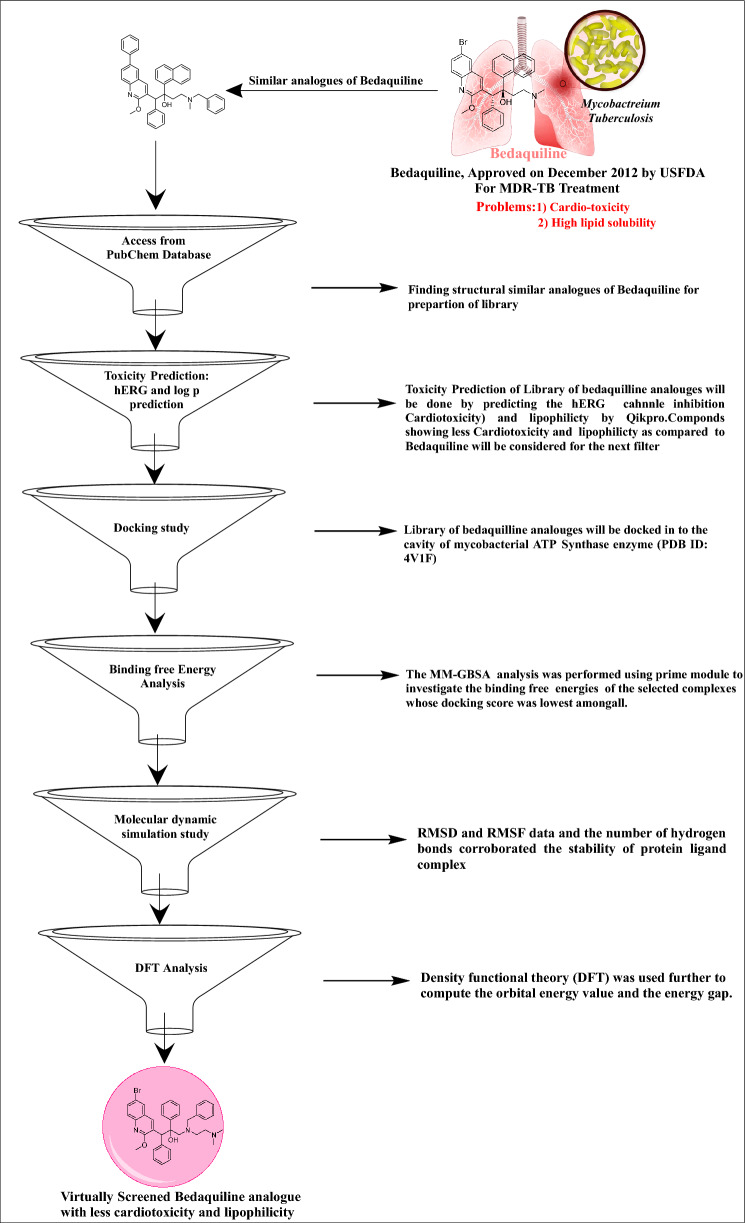
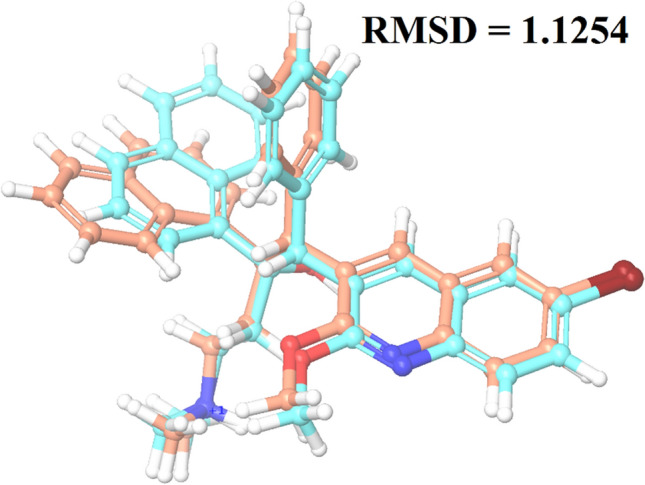
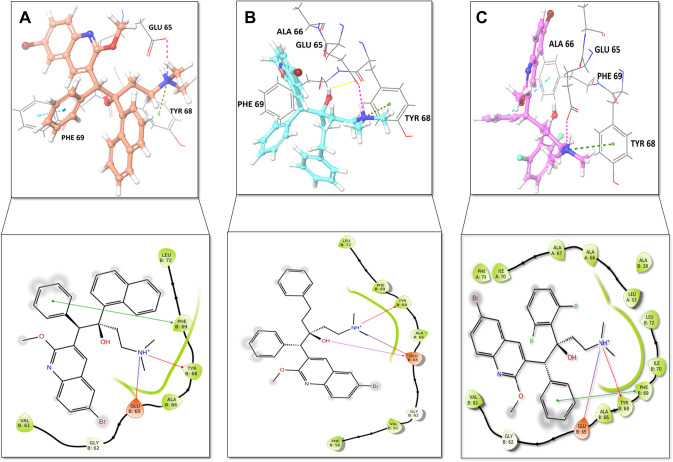
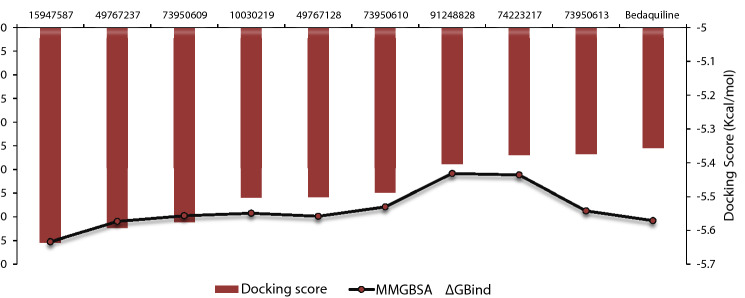
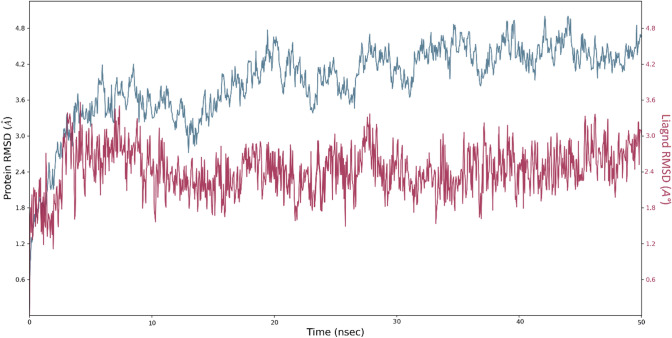
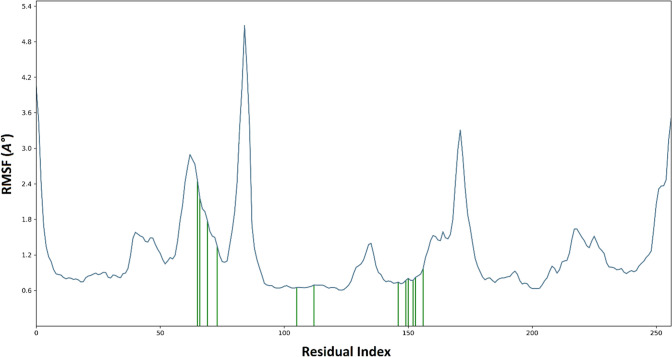
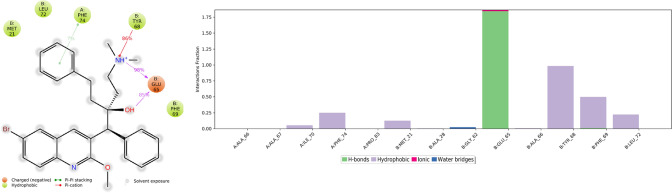
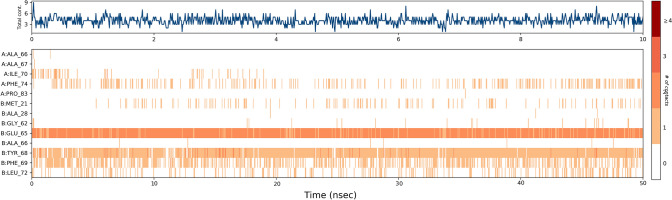
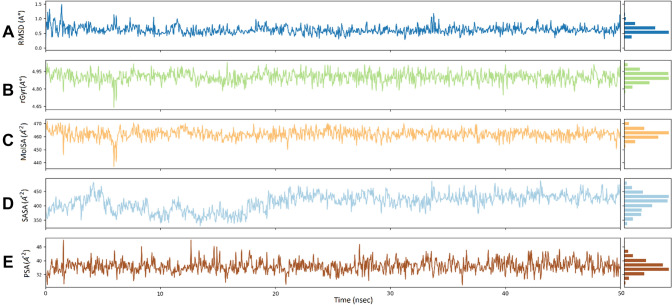
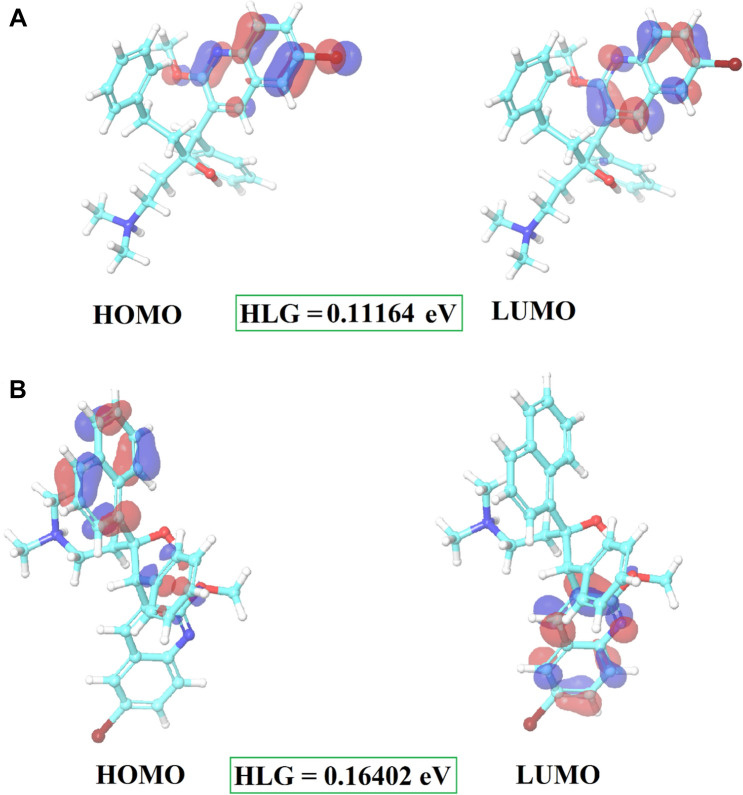
Since the last 4 decades, Bedaquiline has been the first drug discovered as a new kind of anti-tubercular agent and received FDA approval in December 2012 to treat pulmonary multi-drug resistance tuberculosis (MDR-TB). It demonstrates excellent efficacy against MDR-TB by effectively inhibiting mycobacterial ATP synthase. In addition to these apparent assets of Bedaquiline, potential disadvantages of Bedaquiline include inhibition of the hERG (human Ether-à-go-related gene; KCNH2), potassium channel (concurrent risk of cardiac toxicity), and risk of phospholipidosis due to its more lipophilic nature. To assist the effective treatment of MDR-TB, highly active Bedaquiline analogs that display a better safety profile are urgently needed. A structure-based virtual screening approach was used to address the toxicity problems associated with Bedaquiline. Among the virtually screened compound, CID 15947587 had significant docking affinity (- 5.636 kcal/mol) and highest binding free energy (ΔG bind - 85.2703 kcal/mol) towards the Mycobacterial ATP synthase enzyme with insignificant cardiotoxicity and lipophilicity. During MD simulation studies (50 ns), the molecule optimizes its conformation to fit better the active receptor site justifying the binding affinity. The obtained results showed that CID15947587 could be a useful template for further optimizing the MDR-TB inhibitor.
Supplementary information: The online version contains supplementary material available at 10.1007/s40203-021-00086-x.
Keywords: Bedaquiline; Docking; HERG; MD simulation; MMGBSA.
© The Author(s), under exclusive licence to Springer-Verlag GmbH Germany, part of Springer Nature 2021.
Conflict of interest statement
Conflict of interestAll authors declare no actual or potential conflict of interest including any financial, personal, or other relationships with other people or organizations.
Figures
References
-
- (1974) Controlled clinical trial of four short-course (6-month) regimens of chemotherapy for treatment of pulmonary tuberculosis. Lancet 2(7889):1100–1106 - PubMed
-
- (2017) Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2018-4. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2018-4
-
- Amala M, Rajamanikandan S, Prabhu D, Surekha K, Jeyakanthan J. Identification of anti-filarial leads against aspartate semialdehyde dehydrogenase of Wolbachia endosymbiont of Brugia malayi: combined molecular docking and molecular dynamics approaches. J Biomol Struct Dyn. 2019;37(2):394–410. doi: 10.1080/07391102.2018.1427633. - DOI - PubMed
-
- Bhowmick S, AlFaris NA, AlTamimi JZ, AlOthman ZA, Aldayel TS, Wabaidur SM, Islam MA. Screening and analysis of bioactive food compounds for modulating the CDK2 protein for cell cycle arrest: multi-cheminformatics approaches for anticancer therapeutics. J Mol Struct. 2020 doi: 10.1016/j.molstruc.2020.128316. - DOI
-
- Bochevarov AD, Harder E, Hughes TF, Greenwood JR, Braden DA, Philipp DM, Rinaldo D, Hall MD, Zhang J, Friesner RA. Jaguar: a high-performance quantum chemistry software program with strengths in life and materials sciences. Int J Quantum Chem. 2013;113(18):2110–2142. doi: 10.1002/qua.24481. - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
