

Genetic therapies for the first molecular disease
- PMID: 33855970
- PMCID: PMC8262557
- DOI: 10.1172/JCI146394
Genetic therapies for the first molecular disease
Abstract
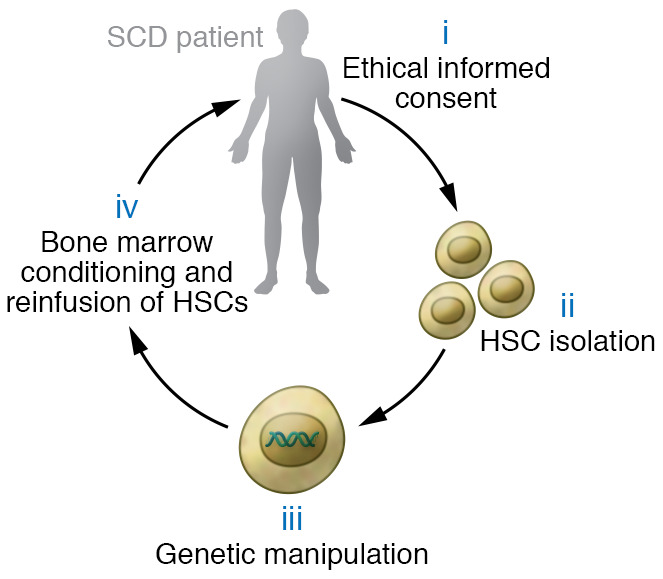
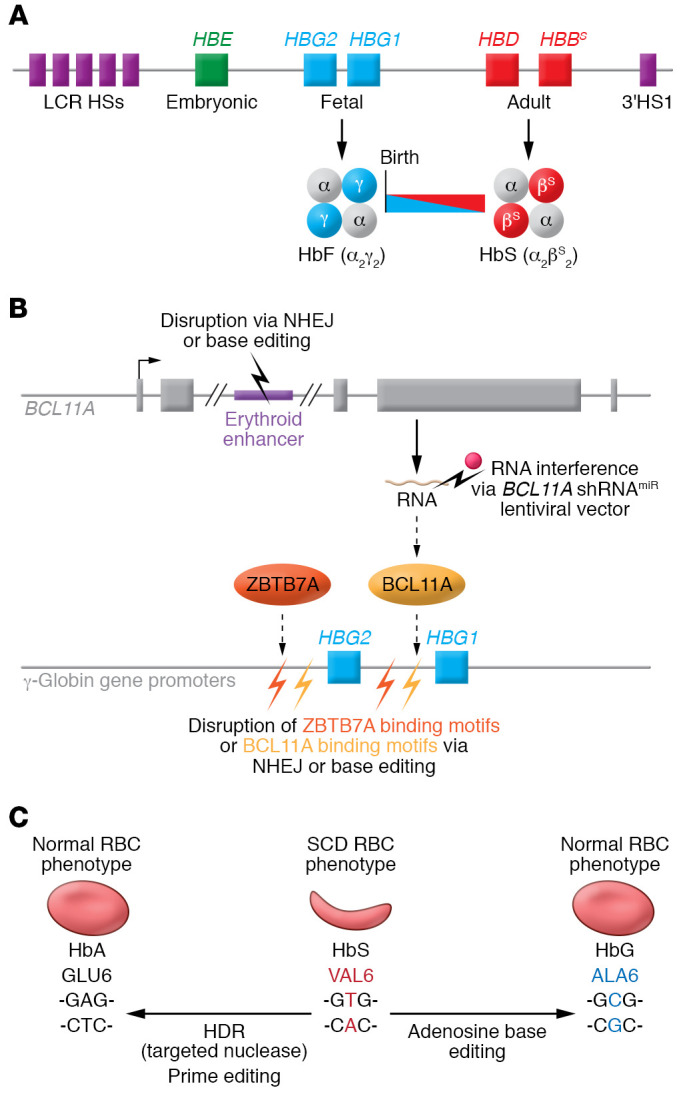
Sickle cell disease (SCD) is a monogenic disorder characterized by recurrent episodes of severe bone pain, multi-organ failure, and early mortality. Although medical progress over the past several decades has improved clinical outcomes and offered cures for many affected individuals living in high-income countries, most SCD patients still experience substantial morbidity and premature death. Emerging technologies to manipulate somatic cell genomes and insights into the mechanisms of developmental globin gene regulation are generating potentially transformative approaches to cure SCD by autologous hematopoietic stem cell (HSC) transplantation. Key components of current approaches include ethical informed consent, isolation of patient HSCs, in vitro genetic modification of HSCs to correct the SCD mutation or circumvent its damaging effects, and reinfusion of the modified HSCs following myelotoxic bone marrow conditioning. Successful integration of these components into effective therapies requires interdisciplinary collaborations between laboratory researchers, clinical caregivers, and patients. Here we summarize current knowledge and research challenges for each key component, emphasizing that the best approaches have yet to be developed.
Conflict of interest statement
Figures

References
-
- Beet EA. The genetics of the sickle-cell trait in a Bantu tribe. Ann Eugen. 1949;14(4):279–284. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
