

Multi-PLI: interpretable multi-task deep learning model for unifying protein-ligand interaction datasets
- PMID: 33858485
- PMCID: PMC8051026
- DOI: 10.1186/s13321-021-00510-6
Multi-PLI: interpretable multi-task deep learning model for unifying protein-ligand interaction datasets
Abstract
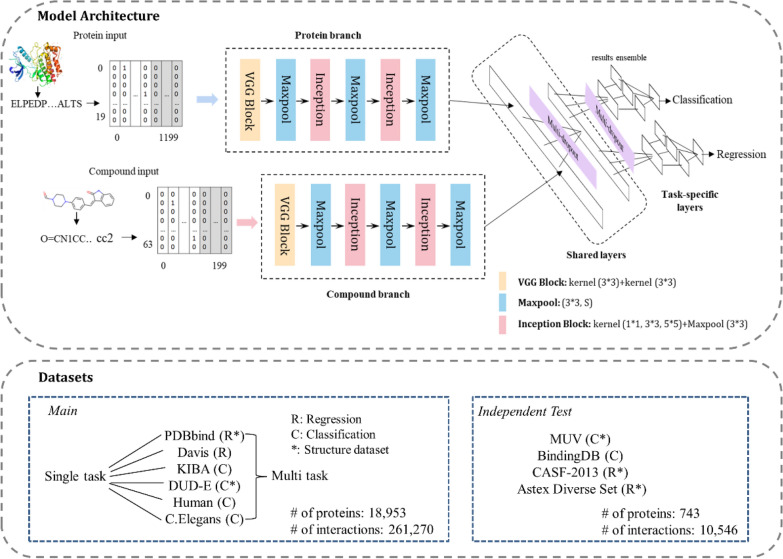
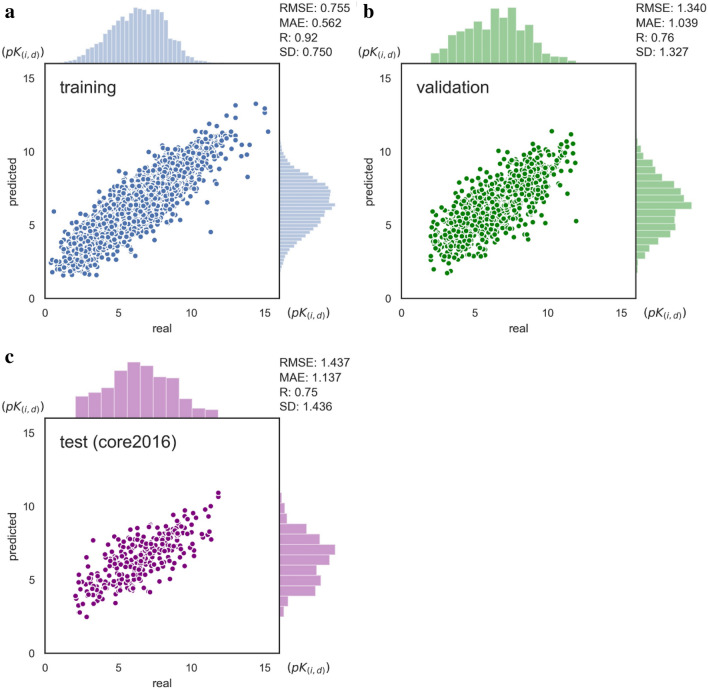
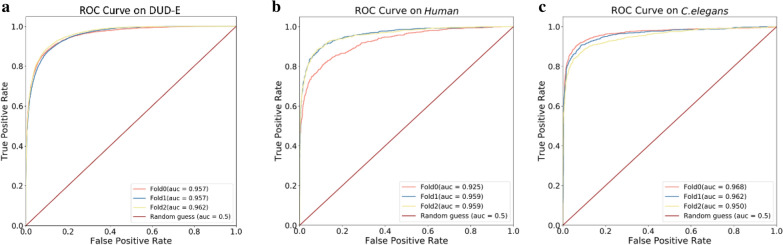
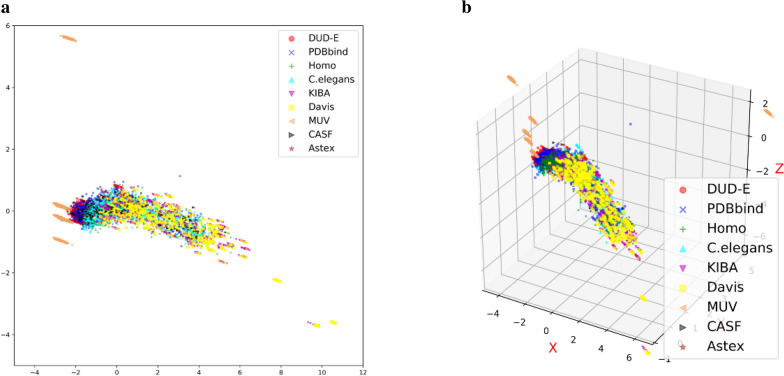
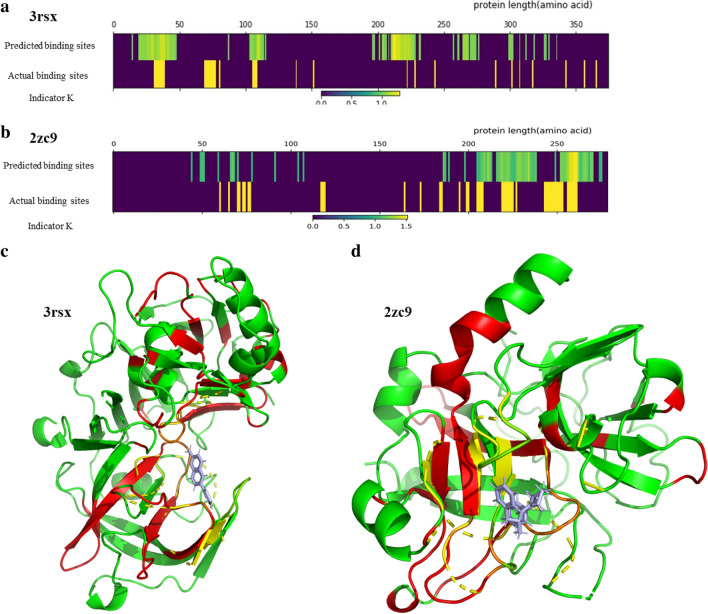
The assessment of protein-ligand interactions is critical at early stage of drug discovery. Computational approaches for efficiently predicting such interactions facilitate drug development. Recently, methods based on deep learning, including structure- and sequence-based models, have achieved impressive performance on several different datasets. However, their application still suffers from a generalizability issue because of insufficient data, especially for structure based models, as well as a heterogeneity problem because of different label measurements and varying proteins across datasets. Here, we present an interpretable multi-task model to evaluate protein-ligand interaction (Multi-PLI). The model can run classification (binding or not) and regression (binding affinity) tasks concurrently by unifying different datasets. The model outperforms traditional docking and machine learning on both binary classification and regression tasks and achieves competitive results compared with some structure-based deep learning methods, even with the same training set size. Furthermore, combined with the proposed occlusion algorithm, the model can predict the important amino acids of proteins that are crucial for binding, thus providing a biological interpretation.
Keywords: Deep learning; Drug discovery; Interpretable; Multi‐task.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Koeppen H, Kriegl J, Lessel U et al (2011) Ligand-based virtual screening. virtual screen princ Challenges, pract Guide 61–85. 10.1002/9783527633326.ch3
-
- Krizhevsky A, Sutskever I, Hinton GE. ImageNet classification with deep convolutional neural networks. Commun ACM. 2017;60:84–90. doi: 10.1145/3065386. - DOI
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
