

Converging genetic and epigenetic drivers of paediatric acute lymphoblastic leukaemia identified by an information-theoretic analysis
- PMID: 33859388
- PMCID: PMC8370714
- DOI: 10.1038/s41551-021-00703-2
Converging genetic and epigenetic drivers of paediatric acute lymphoblastic leukaemia identified by an information-theoretic analysis
Abstract
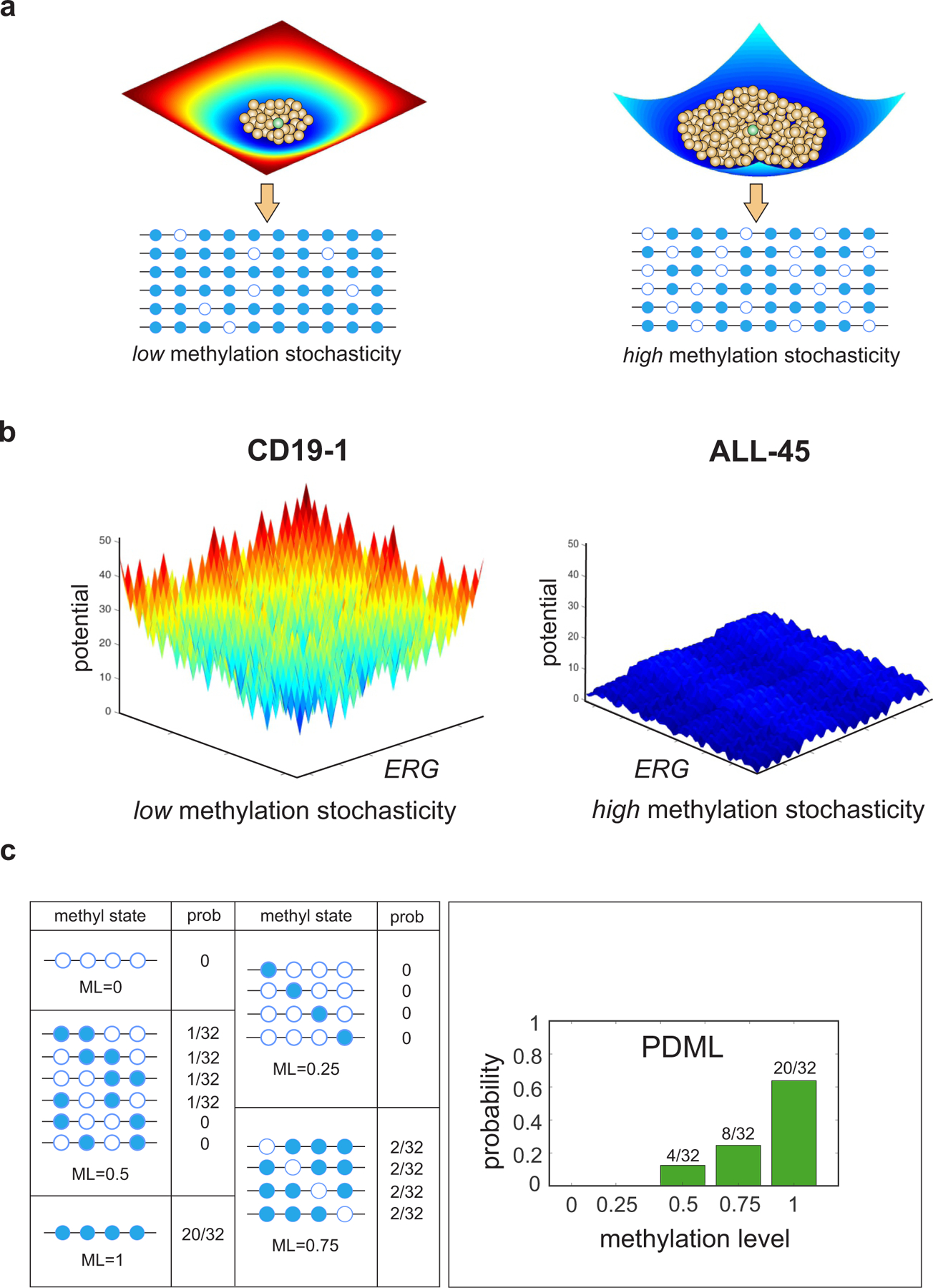
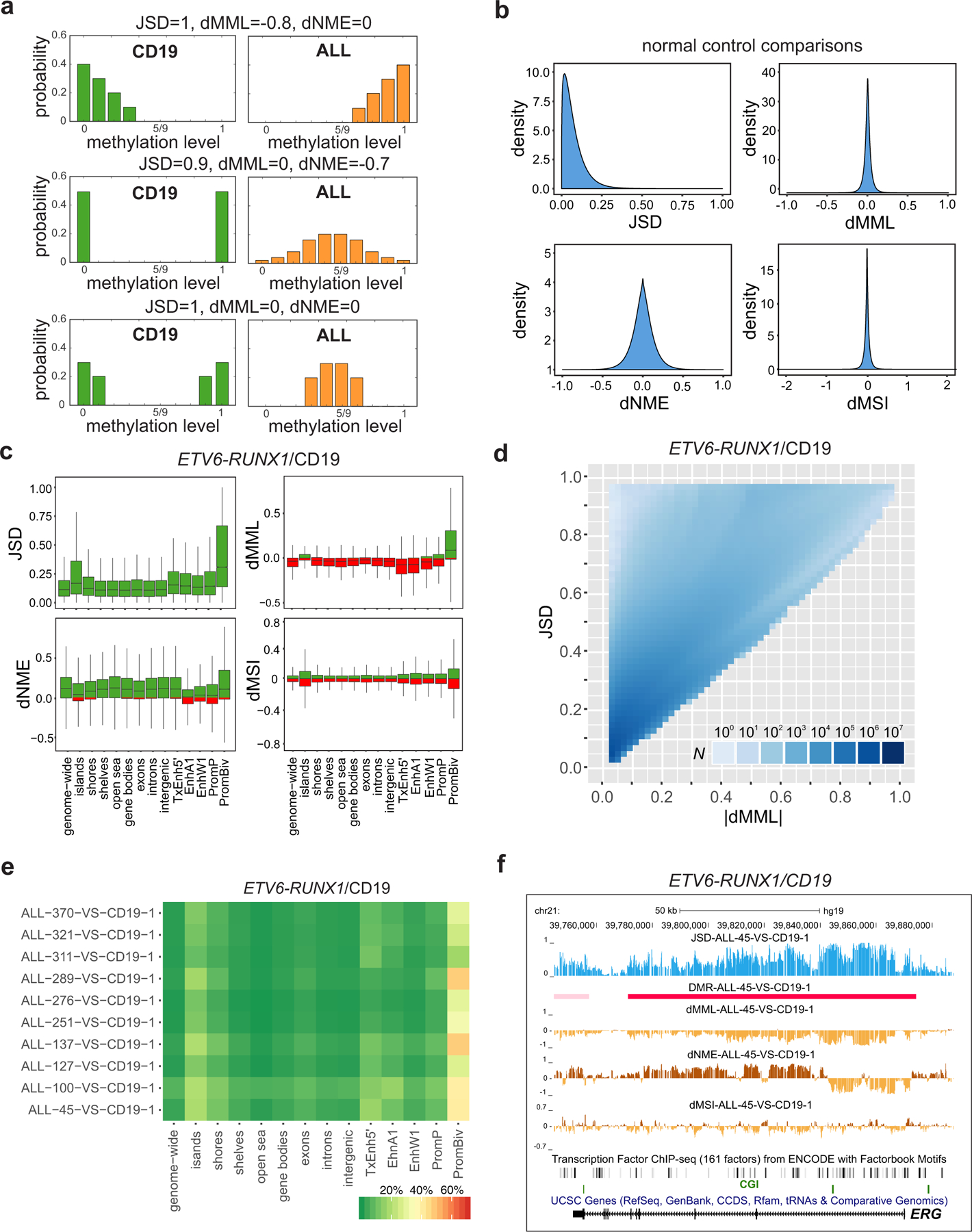
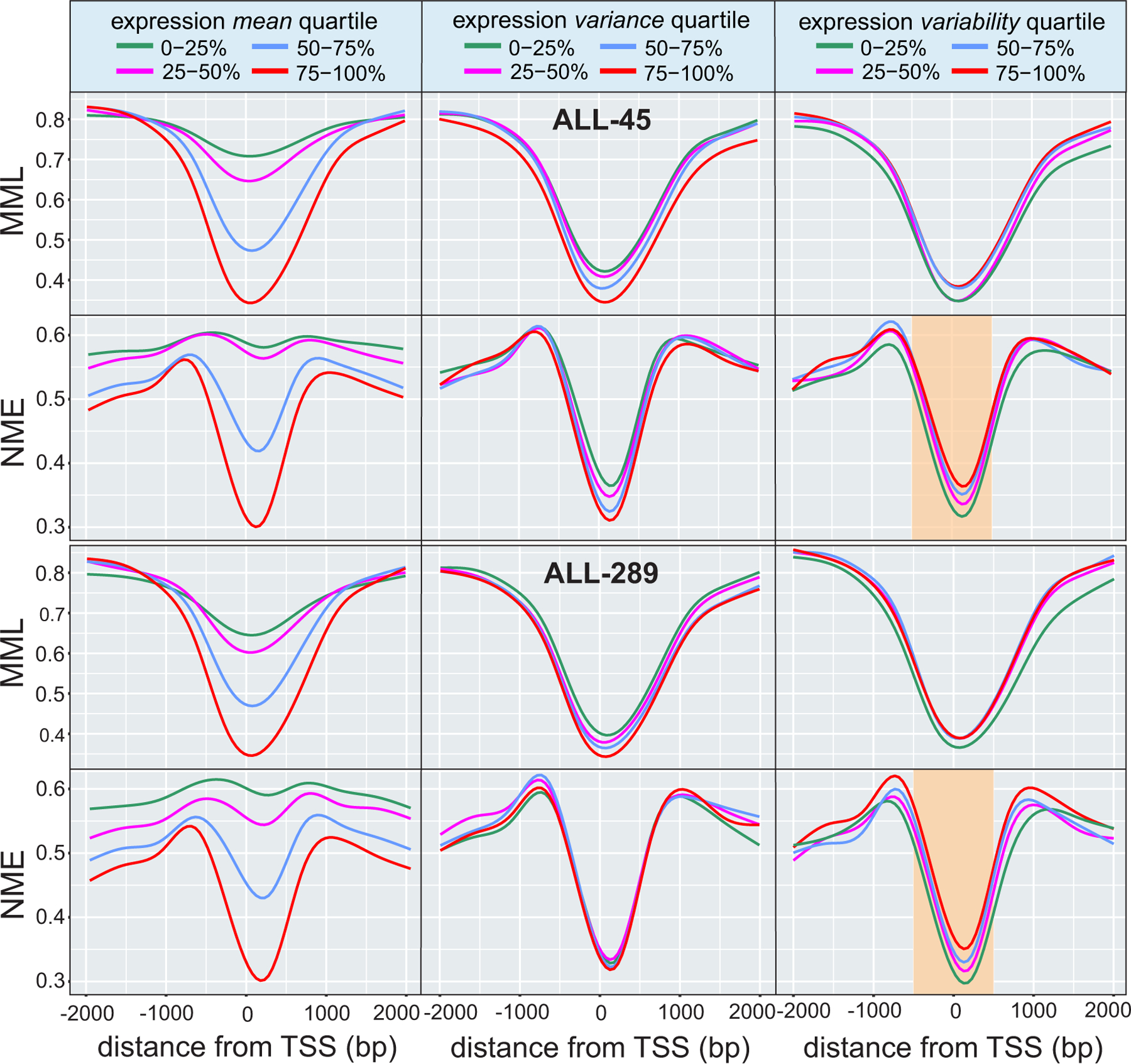
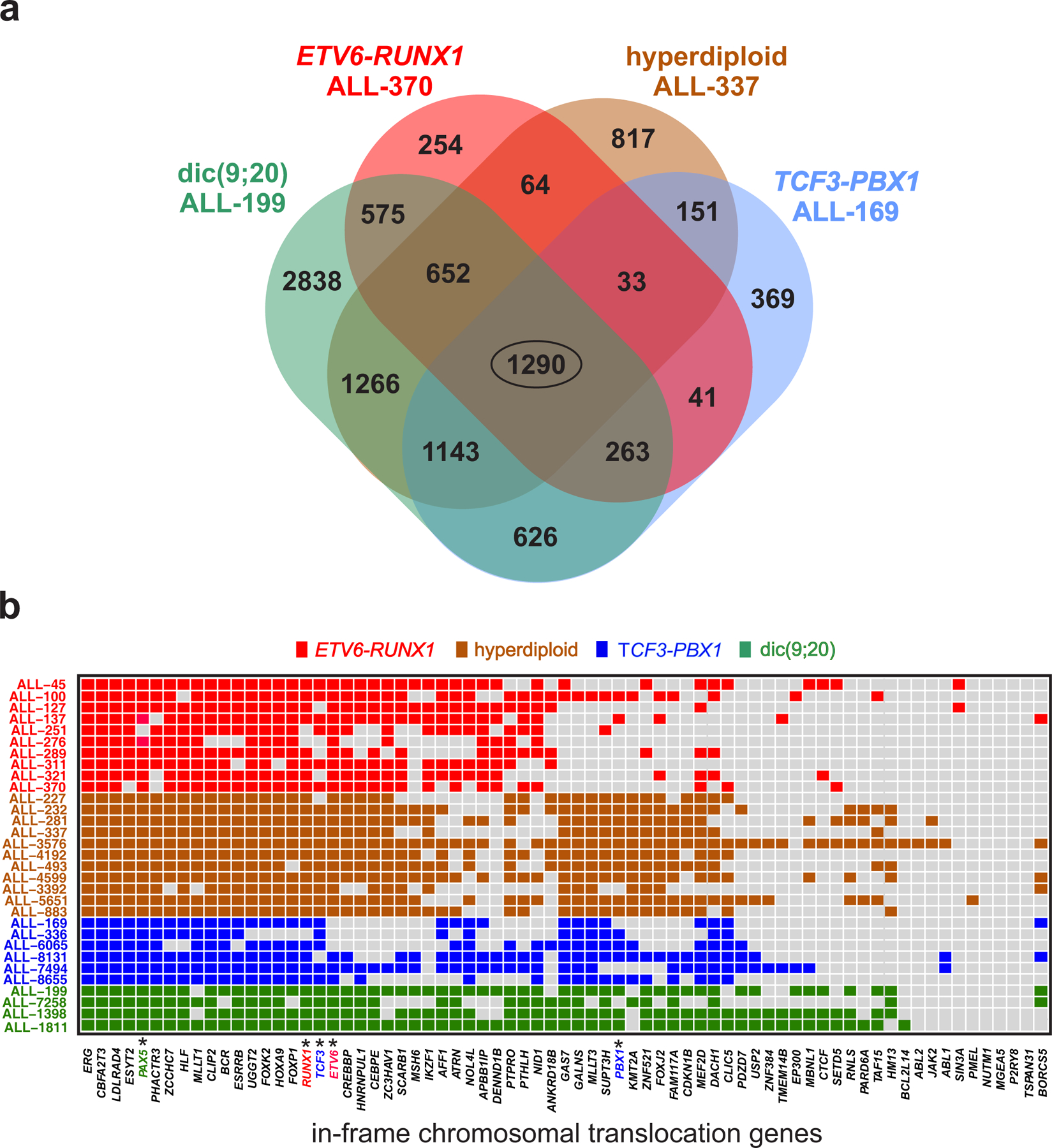
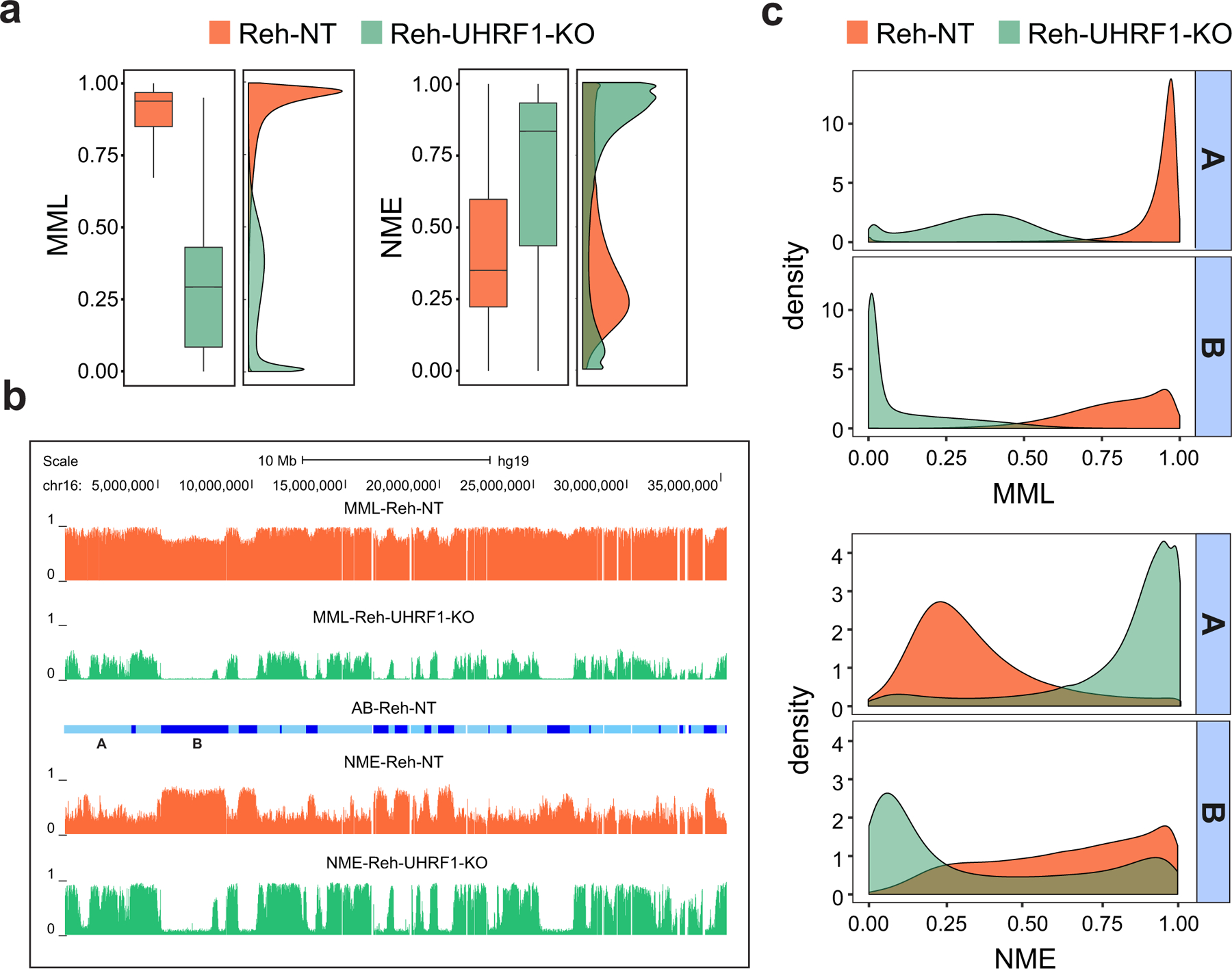
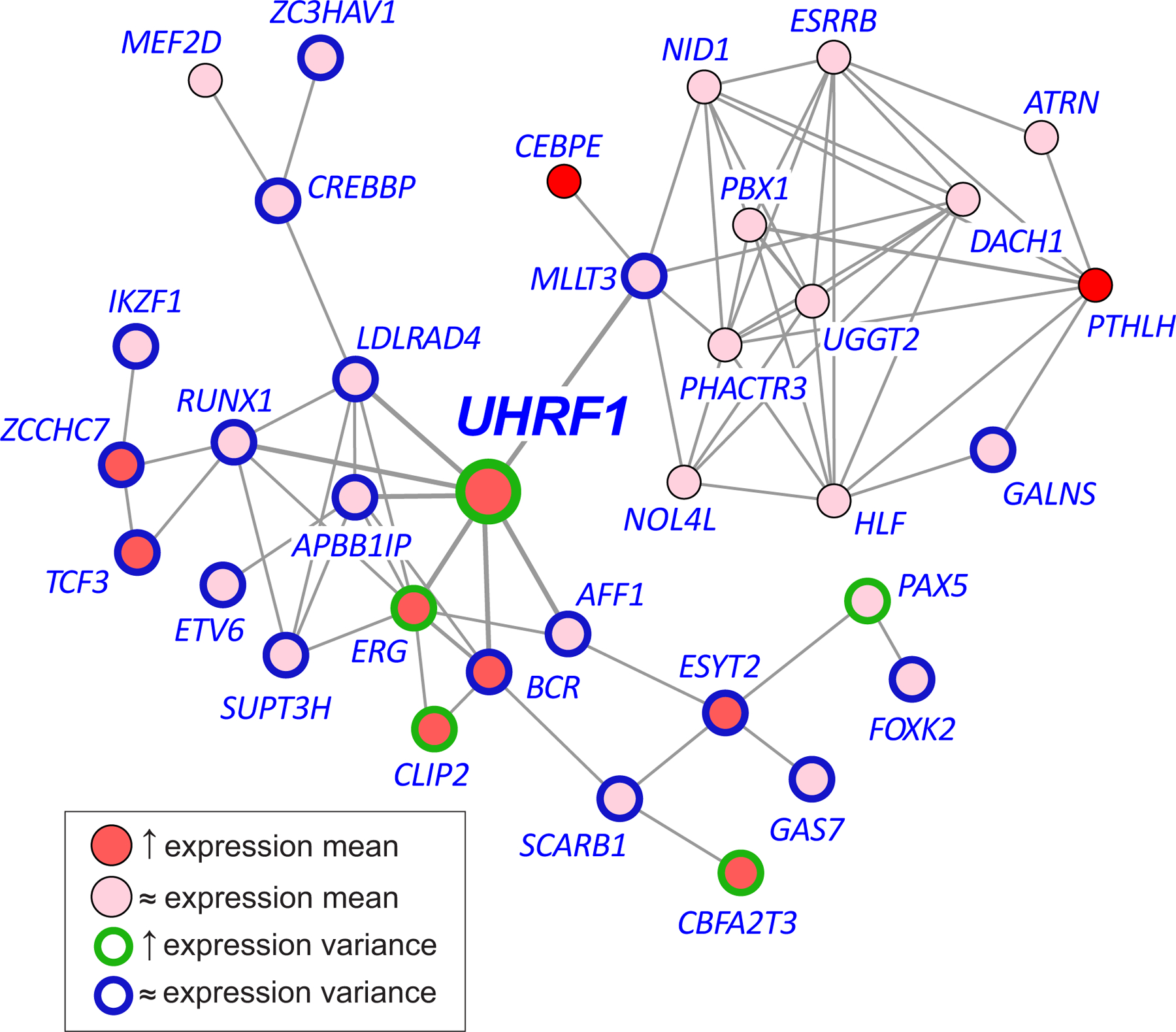
In cancer, linking epigenetic alterations to drivers of transformation has been difficult, in part because DNA methylation analyses must capture epigenetic variability, which is central to tumour heterogeneity and tumour plasticity. Here, by conducting a comprehensive analysis, based on information theory, of differences in methylation stochasticity in samples from patients with paediatric acute lymphoblastic leukaemia (ALL), we show that ALL epigenomes are stochastic and marked by increased methylation entropy at specific regulatory regions and genes. By integrating DNA methylation and single-cell gene-expression data, we arrived at a relationship between methylation entropy and gene-expression variability, and found that epigenetic changes in ALL converge on a shared set of genes that overlap with genetic drivers involved in chromosomal translocations across the disease spectrum. Our findings suggest that an epigenetically driven gene-regulation network, with UHRF1 (ubiquitin-like with PHD and RING finger domains 1) as a central node, links genetic drivers and epigenetic mediators in ALL.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
References
-
- Mullighan CG The molecular genetic makeup of acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program 2012, 389–96 (2012). - PubMed
-
- Hunger SP & Mullighan CG Acute lymphoblastic leukemia in children. N Engl J Med 373, 1541–52 (2015). - PubMed
-
- Grobner SN et al.The landscape of genomic alterations across childhood cancers. Nature 555, 321–327 (2018). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
