

Role of the IL23/IL17 Pathway in Crohn's Disease
- PMID: 33859636
- PMCID: PMC8042267
- DOI: 10.3389/fimmu.2021.622934
Role of the IL23/IL17 Pathway in Crohn's Disease
Abstract
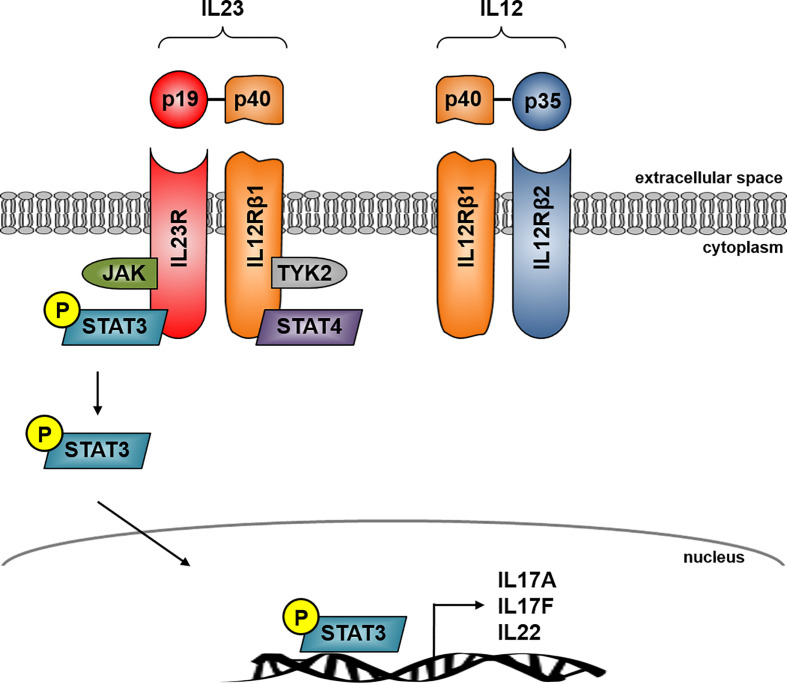
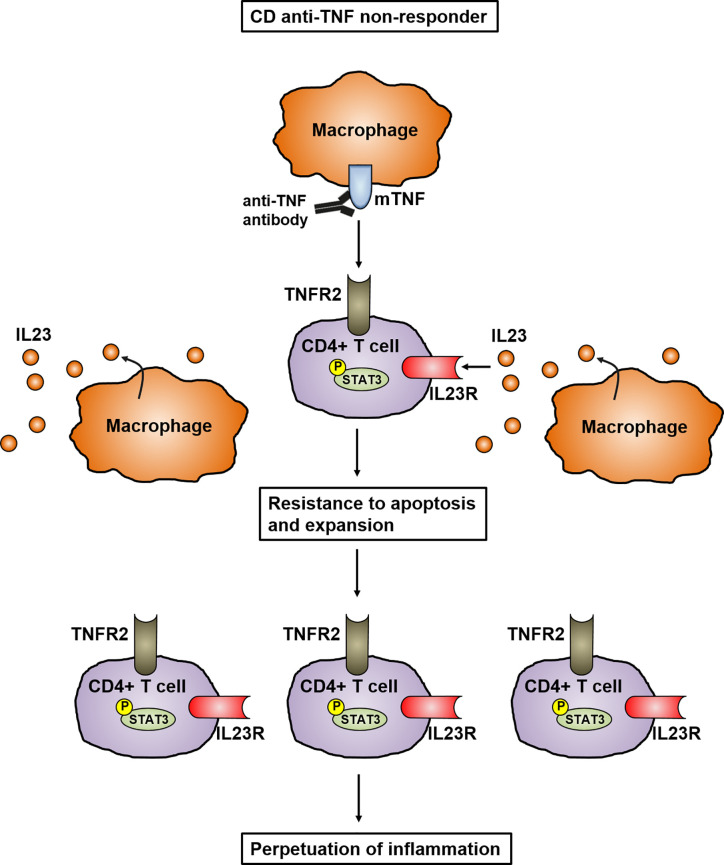
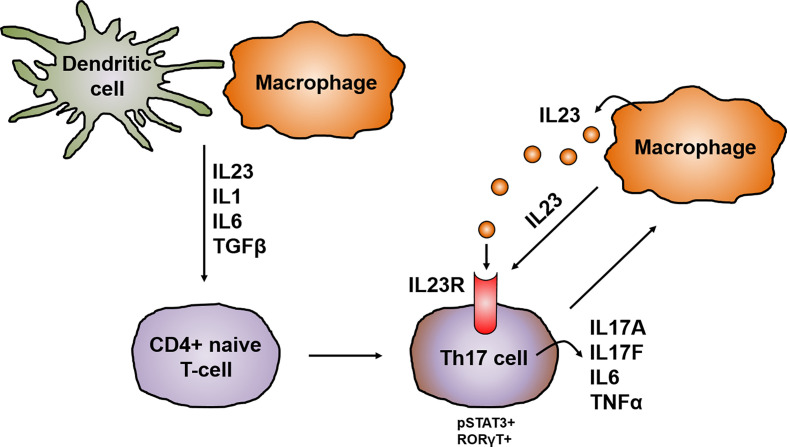
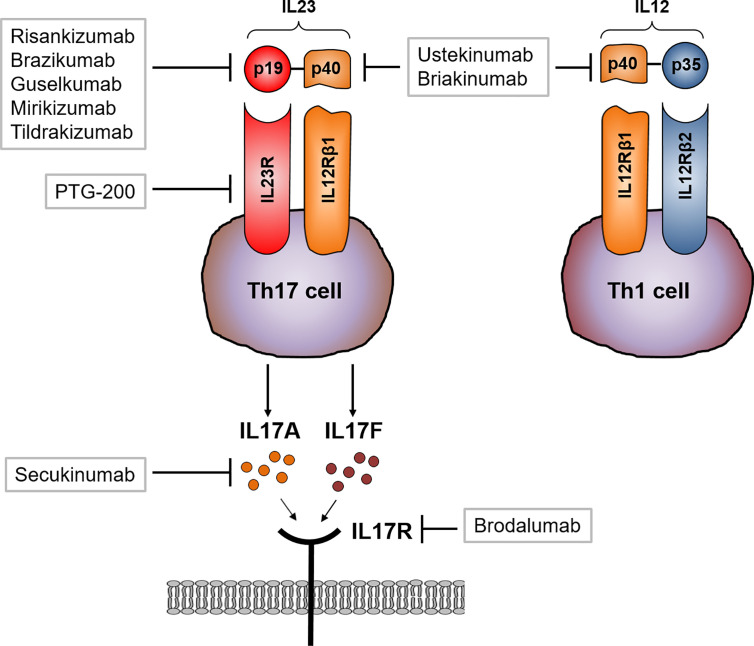
Crohn's disease (CD) is a chronic relapsing disorder of the gastrointestinal tract and represents one of the main entities of inflammatory bowel disease (IBD). CD affects genetically susceptible patients that are influenced by environmental factors and the intestinal microbiome, which results in excessive activation of the mucosal immune system and aberrant cytokine responses. Various studies have implicated the pro-inflammatory cytokines IL17 and IL23 in the pathogenesis of CD. IL23 is a member of the IL12 family of cytokines and is able to enhance and affect the expansion of pathogenic T helper type 17 (Th17) cells through various mechanisms, including maintenance of Th17 signature genes, upregulation of effector genes or suppression of repressive factors. Moreover, IL17 and IL23 signaling is able to induce a cascade of pro-inflammatory molecules like TNF, IFNγ, IL22, lymphotoxin, IL1β and lipopolysaccharide (LPS). Here, IL17A and TNF are known to mediate signaling synergistically to drive expression of inflammatory genes. Recent advances in understanding the immunopathogenetic mechanisms underlying CD have led to the development of new biological therapies that selectively intervene and inhibit inflammatory processes caused by pro-inflammatory mediators like IL17 and IL23. Recently published data demonstrate that treatment with selective IL23 inhibitors lead to markedly high response rates in the cohort of CD patients that failed previous anti-TNF therapy. Macrophages are considered as a main source of IL23 in the intestine and are supposed to play a key role in the molecular crosstalk with T cell subsets and innate lymphoid cells in the gut. The following review focuses on mechanisms, pathways and specific therapies in Crohn's disease underlying the IL23/IL17 pathway.
Keywords: Crohn’s disease; IL17/IL23 axis; anti-TNF therapy; inflammation; intestinal immunity; non-responder; resistance to apoptosis.
Copyright © 2021 Schmitt, Neurath and Atreya.
Conflict of interest statement
RA has served as a speaker, or consultant, or received research grants from AbbVie, Amgen, Arena Pharmaceuticals, Biogen, Boehringer Ingelheim, Celltrion Healthcare, Dr. Falk Pharma, Ferring, Galapagos, Gilead, InDex Pharmaceuticals, Janssen-Cilag, Kliniksa Pharmaceuticals, MSD Sharp & Dohme, Novartis, Pfizer, Roche Pharma, Samsung Bioepsis, Takeda, and Tillotts Pharma. MN reports research grants and/or personal fees from Abbvie, MSD, Takeda, Boehringer, Roche, Pfizer, Janssen, Pentax and PPD. The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical