

Turning cold tumors into hot tumors by improving T-cell infiltration
- PMID: 33859752
- PMCID: PMC8039952
- DOI: 10.7150/thno.58390
Turning cold tumors into hot tumors by improving T-cell infiltration
Abstract
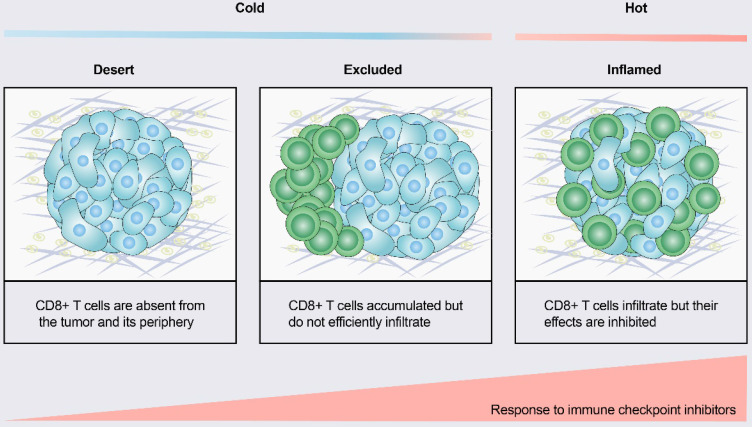
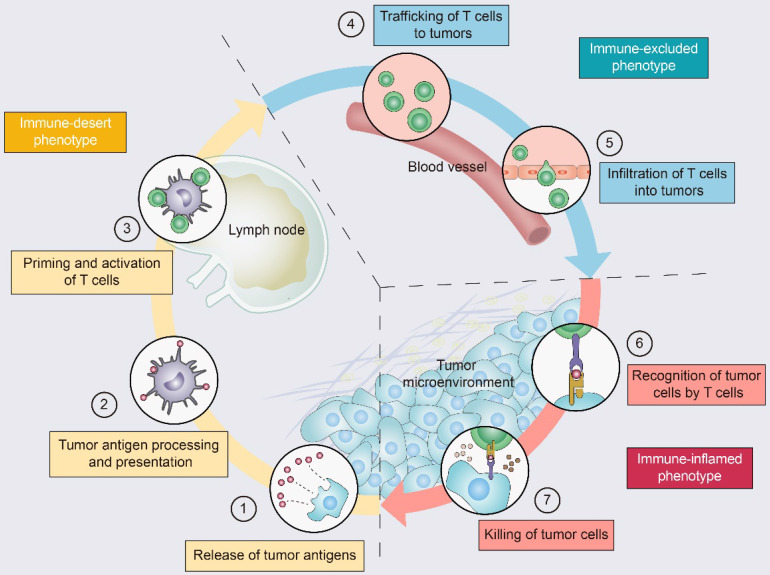
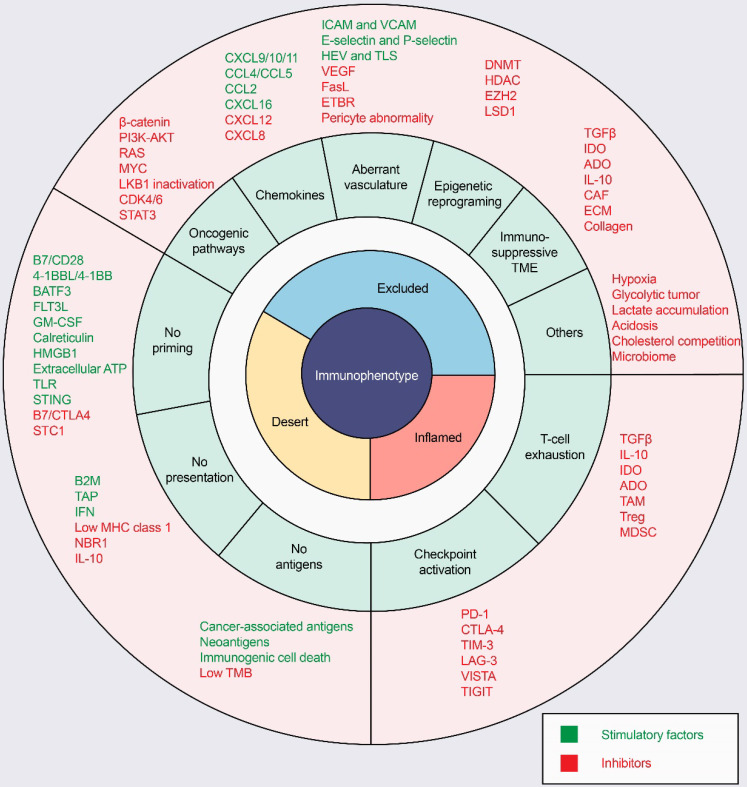
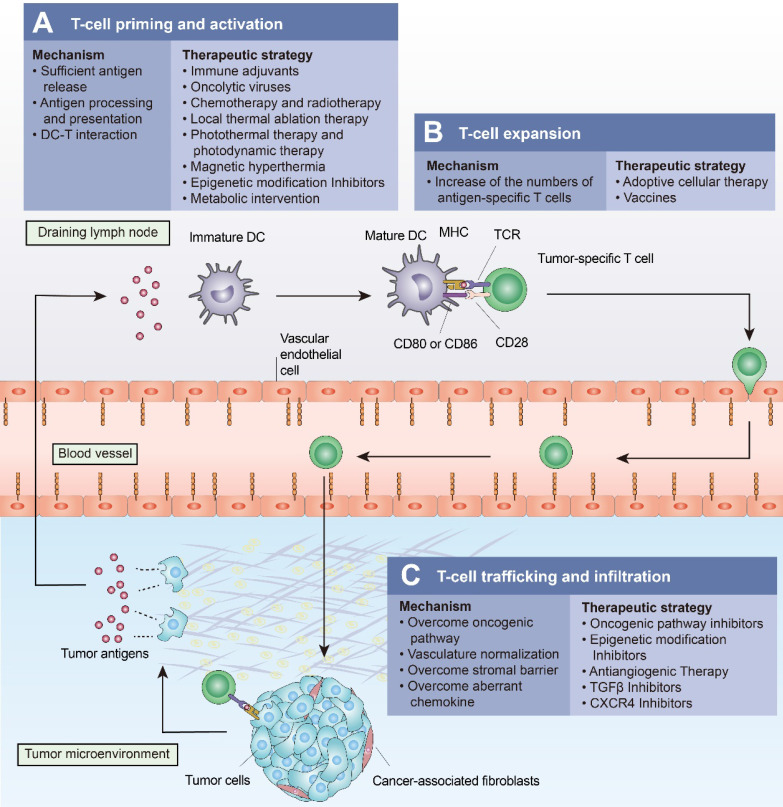
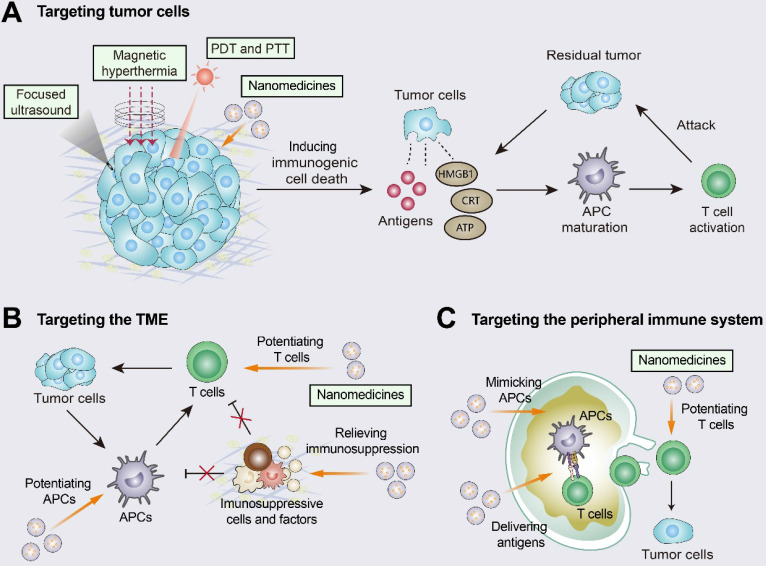
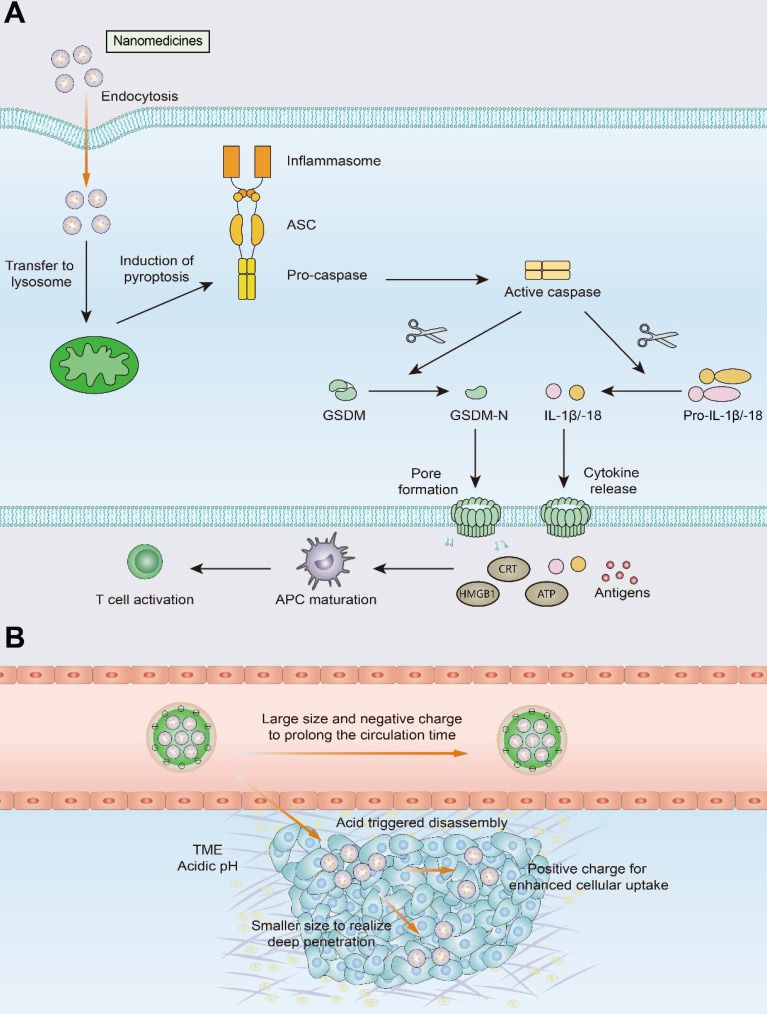
Immunotherapy, represented by immune checkpoint inhibitors (ICIs), has greatly improved the clinical efficacy of malignant tumor therapy. ICI-mediated antitumor responses depend on the infiltration of T cells capable of recognizing and killing tumor cells. ICIs are not effective in "cold tumors", which are characterized by the lack of T-cell infiltration. To realize the full potential of immunotherapy and solve this obstacle, it is essential to understand the drivers of T-cell infiltration into tumors. We present a critical review of our understanding of the mechanisms underlying "cold tumors", including impaired T-cell priming and deficient T-cell homing to tumor beds. "Hot tumors" with significant T-cell infiltration are associated with better ICI efficacy. In this review, we summarize multiple strategies that promote the transformation of "cold tumors" into "hot tumors" and discuss the mechanisms by which these strategies lead to increased T-cell infiltration. Finally, we discuss the application of nanomaterials to tumor immunotherapy and provide an outlook on the future of this emerging field. The combination of nanomedicines and immunotherapy enhances cross-presentation of tumor antigens and promotes T-cell priming and infiltration. A deeper understanding of these mechanisms opens new possibilities for the development of multiple T cell-based combination therapies to improve ICI effectiveness.
Keywords: T-cell infiltration; T-cell priming; cold tumor; immune checkpoint inhibitors; nanomedicine.
© The author(s).
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures
References
-
- Garon E, Rizvi N, Hui R, Leighl N, Balmanoukian A, Eder J. et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–28. - PubMed
-
- Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–30. - PubMed
-
- Bruni D, Angell H, Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer. 2020;20:662–80. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
