

Identification of long regulatory elements in the genome of Plasmodium falciparum and other eukaryotes
- PMID: 33861755
- PMCID: PMC8081344
- DOI: 10.1371/journal.pcbi.1008909
Identification of long regulatory elements in the genome of Plasmodium falciparum and other eukaryotes
Abstract
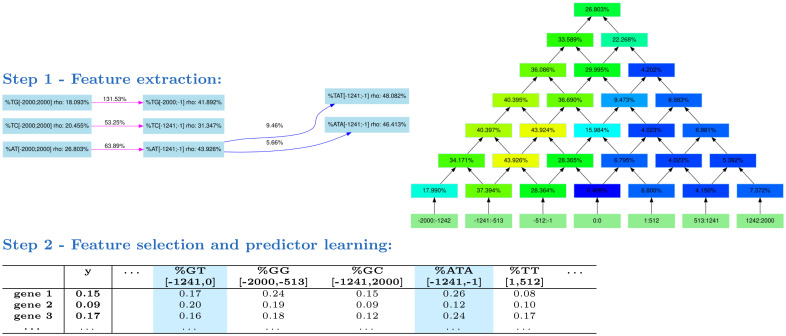
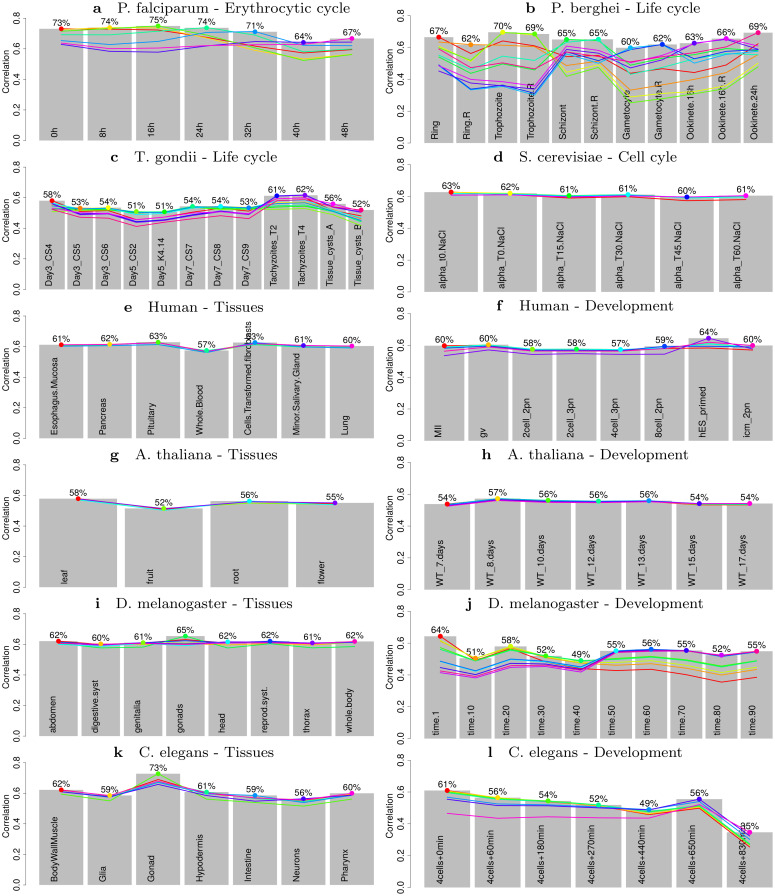
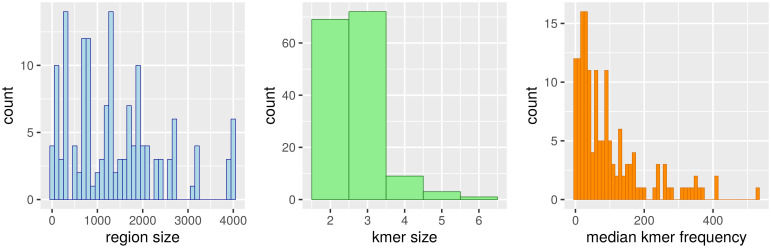
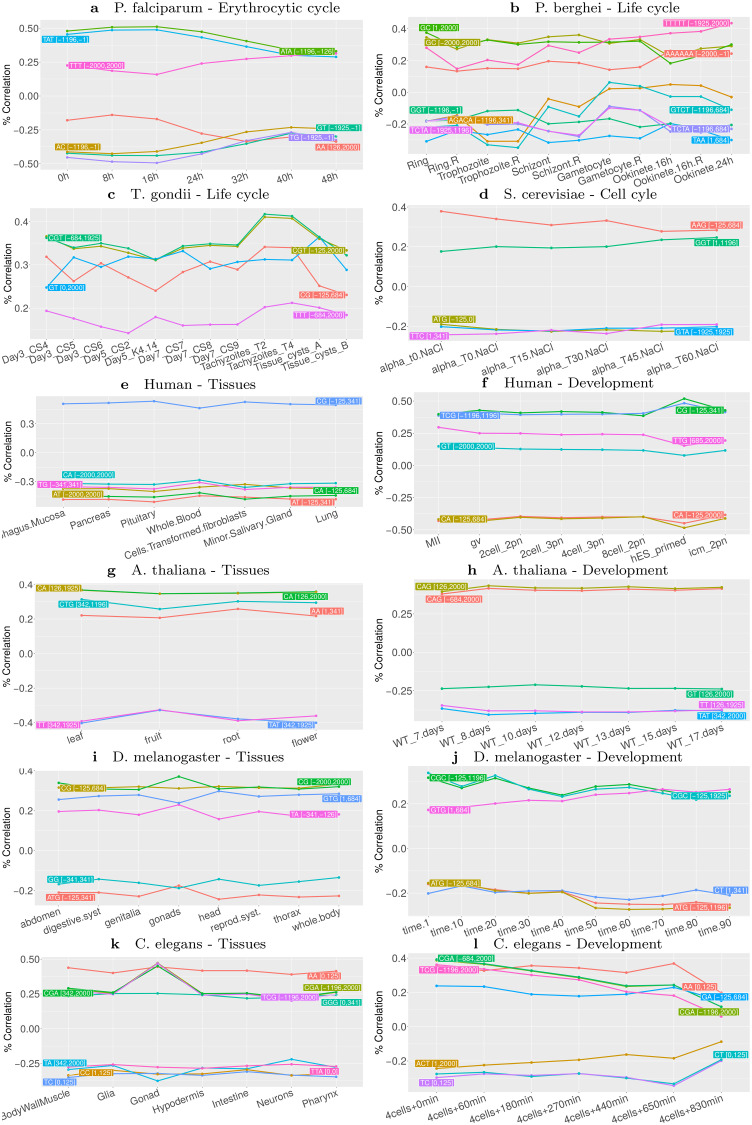
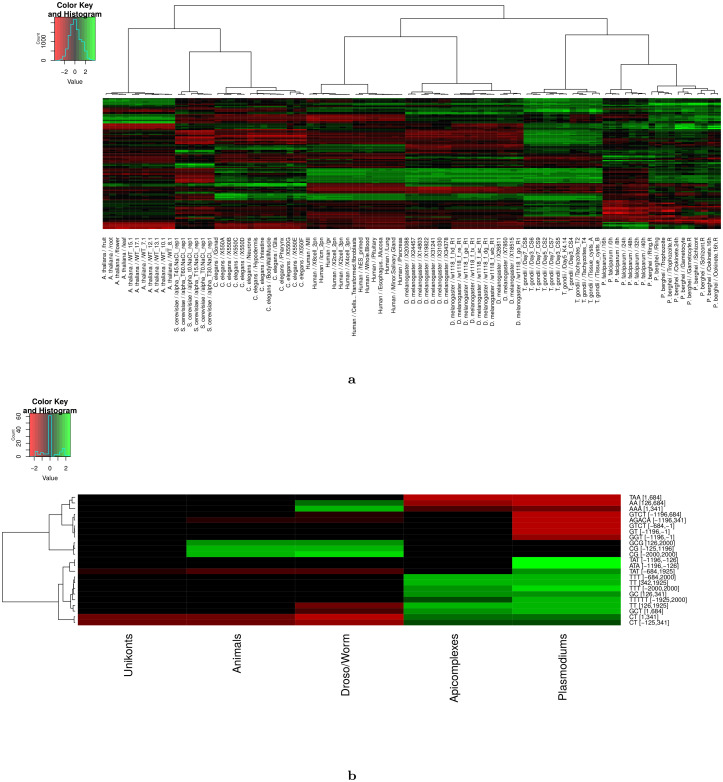
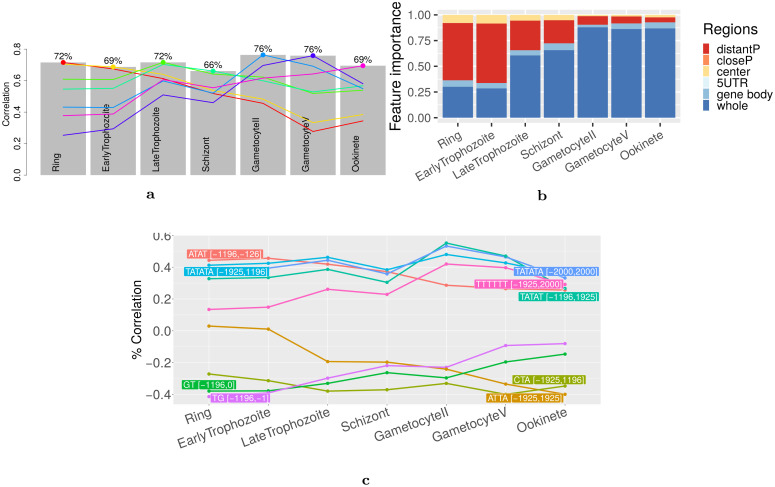
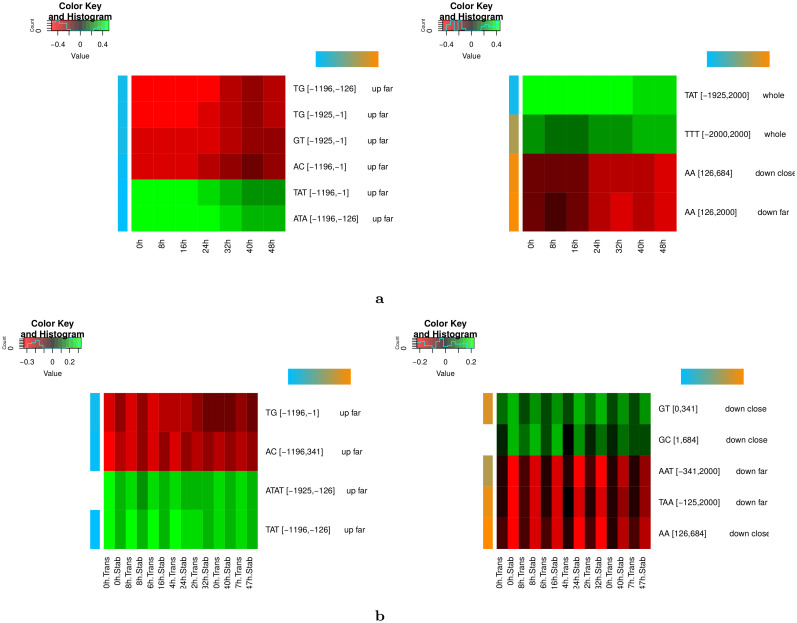
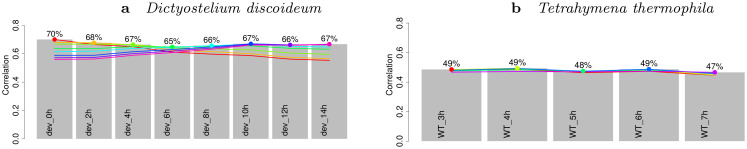
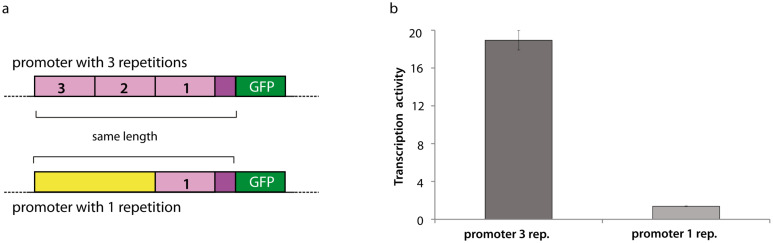
Long regulatory elements (LREs), such as CpG islands, polydA:dT tracts or AU-rich elements, are thought to play key roles in gene regulation but, as opposed to conventional binding sites of transcription factors, few methods have been proposed to formally and automatically characterize them. We present here a computational approach named DExTER (Domain Exploration To Explain gene Regulation) dedicated to the identification of candidate LREs (cLREs) and apply it to the analysis of the genomes of P. falciparum and other eukaryotes. Our analyses show that all tested genomes contain several cLREs that are somewhat conserved along evolution, and that gene expression can be predicted with surprising accuracy on the basis of these long regions only. Regulation by cLREs exhibits very different behaviours depending on species and conditions. In P. falciparum and other Apicomplexan organisms as well as in Dictyostelium discoideum, the process appears highly dynamic, with different cLREs involved at different phases of the life cycle. For multicellular organisms, the same cLREs are involved in all tissues, but a dynamic behavior is observed along embryonic development stages. In P. falciparum, whose genome is known to be strongly depleted of transcription factors, cLREs are predictive of expression with an accuracy above 70%, and our analyses show that they are associated with both transcriptional and post-transcriptional regulation signals. Moreover, we assessed the biological relevance of one LRE discovered by DExTER in P. falciparum using an in vivo reporter assay. The source code (python) of DExTER is available at https://gite.lirmm.fr/menichelli/DExTER.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Similar articles
-
In silico discovery of transcription regulatory elements in Plasmodium falciparum.BMC Genomics. 2008 Feb 7;9:70. doi: 10.1186/1471-2164-9-70. BMC Genomics. 2008. PMID: 18257930 Free PMC article.
-
Identification of regulatory elements in the Plasmodium falciparum genome.Mol Biochem Parasitol. 2004 Mar;134(1):75-88. doi: 10.1016/j.molbiopara.2003.11.004. Mol Biochem Parasitol. 2004. PMID: 14747145
-
Analysis of nucleosome positioning landscapes enables gene discovery in the human malaria parasite Plasmodium falciparum.BMC Genomics. 2015 Nov 25;16:1005. doi: 10.1186/s12864-015-2214-9. BMC Genomics. 2015. PMID: 26607328 Free PMC article.
-
Combinatorial gene regulation in Plasmodium falciparum.Trends Genet. 2006 Feb;22(2):73-8. doi: 10.1016/j.tig.2005.12.002. Epub 2005 Dec 27. Trends Genet. 2006. PMID: 16380193 Review.
-
Epigenetic Players of Chromatin Structure Regulation in Plasmodium falciparum.Chembiochem. 2019 May 15;20(10):1225-1230. doi: 10.1002/cbic.201800718. Epub 2019 Mar 4. Chembiochem. 2019. PMID: 30632244 Review.
Cited by
-
TFscope: systematic analysis of the sequence features involved in the binding preferences of transcription factors.Genome Biol. 2024 Jul 10;25(1):187. doi: 10.1186/s13059-024-03321-8. Genome Biol. 2024. PMID: 38987807 Free PMC article.
-
Advancing Regulatory Genomics With Machine Learning.Bioinform Biol Insights. 2024 Dec 24;18:11779322241249562. doi: 10.1177/11779322241249562. eCollection 2024. Bioinform Biol Insights. 2024. PMID: 39735654 Free PMC article. Review.
References
-
- Toenhake CG, Fraschka SAK, Vijayabaskar MS, Westhead DR, van Heeringen SJ, Bártfai R. Chromatin Accessibility-Based Characterization of the Gene Regulatory Network Underlying Plasmodium falciparum Blood-Stage Development. Cell Host & Microbe. 2018. April;23(4):557–569.e9. 10.1016/j.chom.2018.03.007 - DOI - PMC - PubMed
-
- Modrzynska K, Pfander C, Chappell L, Yu L, Suarez C, Dundas K, et al.. A Knockout Screen of ApiAP2 Genes Reveals Networks of Interacting Transcriptional Regulators Controlling the Plasmodium Life Cycle. Cell Host & Microbe. 2017. January;21(1):11–22. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5241200/. 10.1016/j.chom.2016.12.003 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
