

Emergence and rapid transmission of SARS-CoV-2 B.1.1.7 in the United States
- PMID: 33861950
- PMCID: PMC8009040
- DOI: 10.1016/j.cell.2021.03.052
Emergence and rapid transmission of SARS-CoV-2 B.1.1.7 in the United States
Abstract
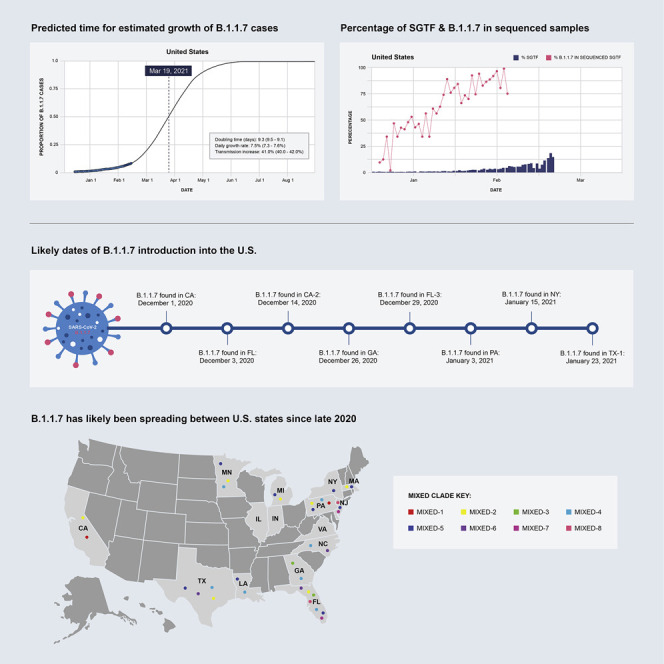
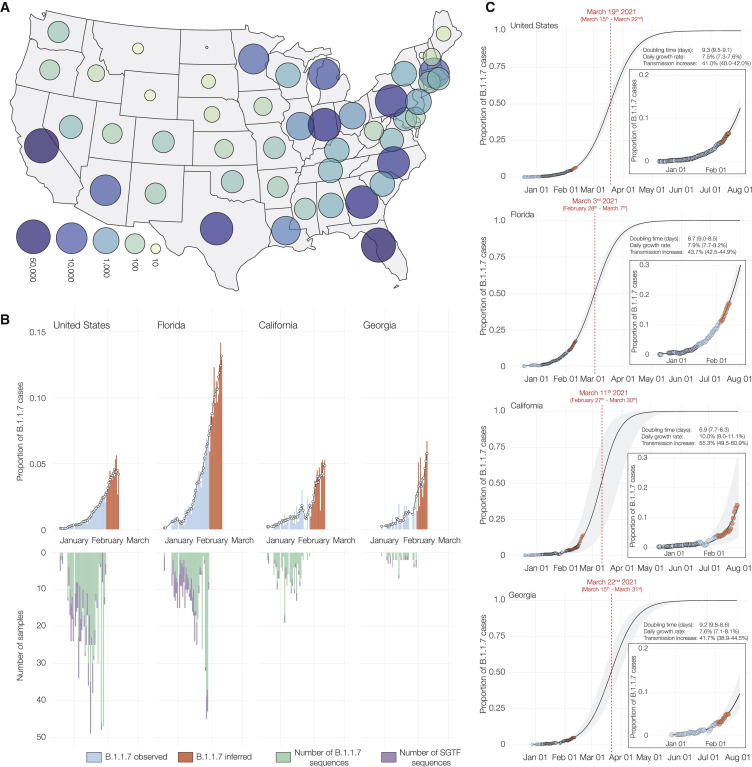
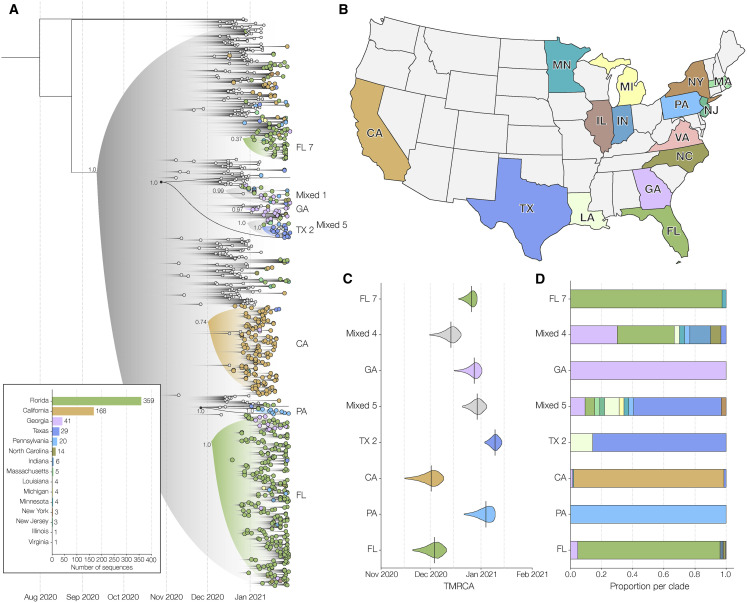
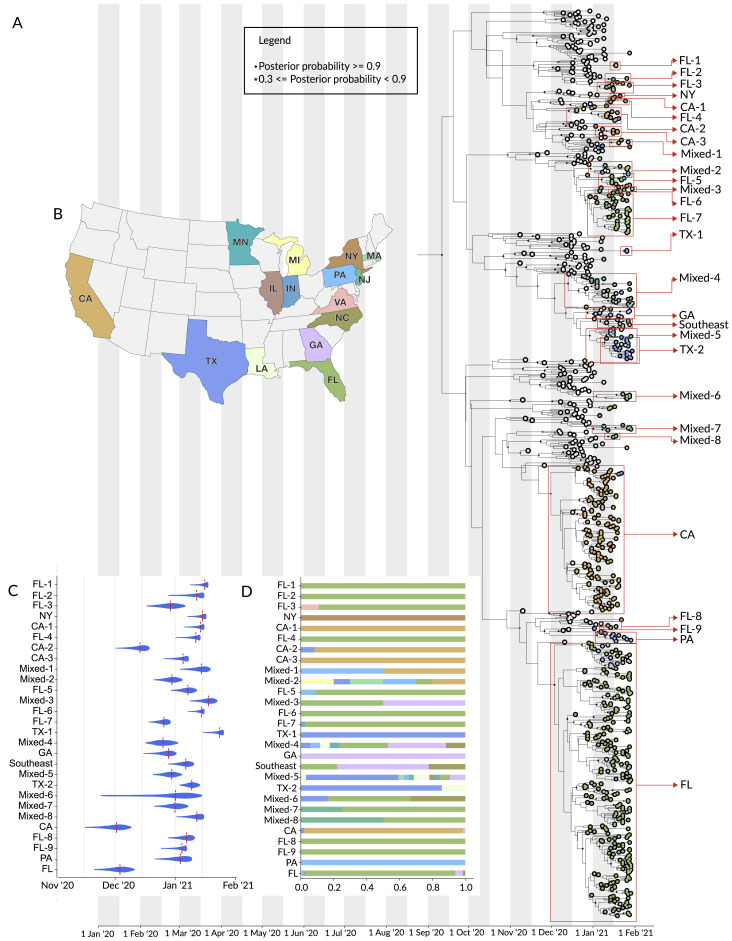
The highly transmissible B.1.1.7 variant of SARS-CoV-2, first identified in the United Kingdom, has gained a foothold across the world. Using S gene target failure (SGTF) and SARS-CoV-2 genomic sequencing, we investigated the prevalence and dynamics of this variant in the United States (US), tracking it back to its early emergence. We found that, while the fraction of B.1.1.7 varied by state, the variant increased at a logistic rate with a roughly weekly doubling rate and an increased transmission of 40%-50%. We revealed several independent introductions of B.1.1.7 into the US as early as late November 2020, with community transmission spreading it to most states within months. We show that the US is on a similar trajectory as other countries where B.1.1.7 became dominant, requiring immediate and decisive action to minimize COVID-19 morbidity and mortality.
Keywords: 501Y.V1; B.1.1.7; COVID-19; SARS-CoV-2; VOC-202012/01; genomic epidemiology; variant of concern.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests N.L.W., A.B., E.T.C., K.M.S.B., S.W., C.R.-G., E. Sandoval, T.C., X.W., J.N., J.M.R., G.L., D.W., D.B., M.L., M.I., S.J., J.T.L., and W.L. are employees of Helix. K. Gietzen, B.S., J.A., K.H., J.L., E.d.F., and P.G.F. are employees of Illumina. J.N., C.R.-G., and M.L. own stock in ILMN. K.G.A. has received consulting fees for advising on SARS-CoV-2, variants, and the COVID-19 pandemic.
Figures
Update of
-
Genomic epidemiology identifies emergence and rapid transmission of SARS-CoV-2 B.1.1.7 in the United States.medRxiv [Preprint]. 2021 Feb 7:2021.02.06.21251159. doi: 10.1101/2021.02.06.21251159. medRxiv. 2021. Update in: Cell. 2021 May 13;184(10):2587-2594.e7. doi: 10.1016/j.cell.2021.03.052. PMID: 33564780 Free PMC article. Updated. Preprint.
Comment in
-
Tracking self-performance in the prefrontal cortex: It's layered.Cell. 2021 May 13;184(10):2534-2536. doi: 10.1016/j.cell.2021.04.030. Cell. 2021. PMID: 33989547 Free PMC article.
References
-
- Ayres D.L., Cummings M.P., Baele G., Darling A.E., Lewis P.O., Swofford D.L., Huelsenbeck J.P., Lemey P., Rambaut A., Suchard M.A. BEAGLE 3: Improved Performance, Scaling, and Usability for a High-Performance Computing Library for Statistical Phylogenetics. Syst. Biol. 2019;68:1052–1061. - PMC - PubMed
-
- Bal A., Destras G., Gaymard A., Regue H., Semanas Q., d’Aubarede C., Billaud G., Laurent F., Gonzalez C., Valette M., et al. Two-step strategy for the identification of SARS-CoV-2 variants co-occurring with spike deletion H69-V70, Lyon, France, August to December 2020. medRxiv. 2020 doi: 10.1101/2020.11.10.20228528. - DOI - PMC - PubMed
-
- Borges B., Sousa C., Menezes L., Gonçalves A.M., Picão M., Almeida J.P., Vieita M., Santos R., Silva A.R., Costa M., et al. 2021. Tracking SARS-CoV-2 VOC 202012/01 (lineage B.1.1.7) dissemination in Portugal: insights from nationwide RT-PCR Spike gene drop out data.https://virological.org/t/tracking-sars-cov-2-voc-202012-01-lineage-b-1-... - PMC - PubMed
-
- CDC . 2021. US COVID-19 Cases Caused by Variants.https://www.cdc.gov/coronavirus/2019-ncov/transmission/variant-cases.html
-
- Chand M., Hopkins S., Dabrera G., Achison C., Barclay W., Ferguson N., Volz E., Loman N., Rambaut A., Barrett J. Public Health England; 2020. Investigation of novel SARS-CoV-2 variant Variant of Concern 202012/01.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
