

On the origin and evolution of SARS-CoV-2
- PMID: 33864026
- PMCID: PMC8050477
- DOI: 10.1038/s12276-021-00604-z
On the origin and evolution of SARS-CoV-2
Abstract
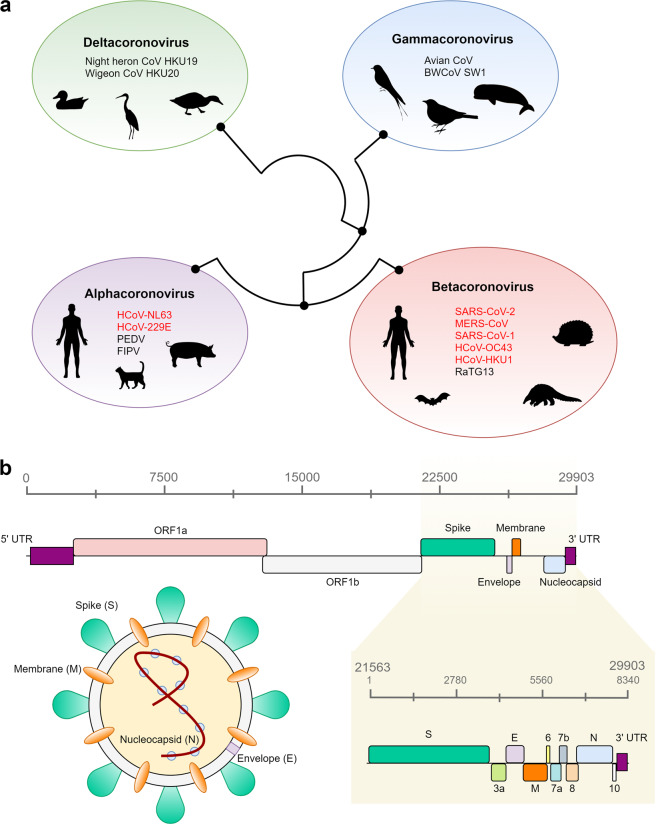
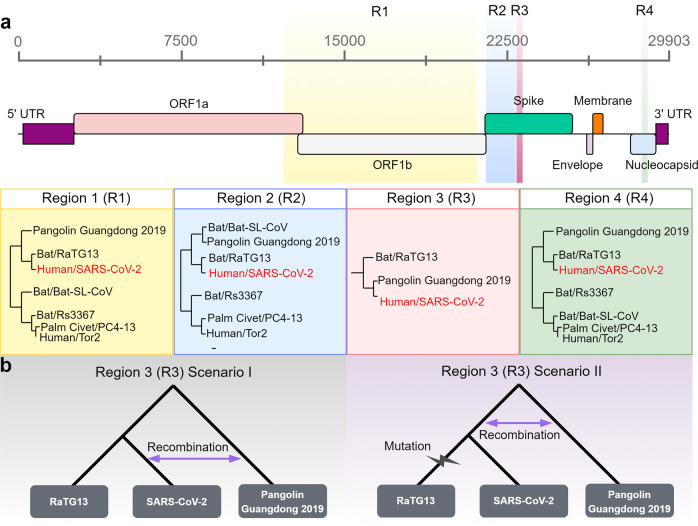
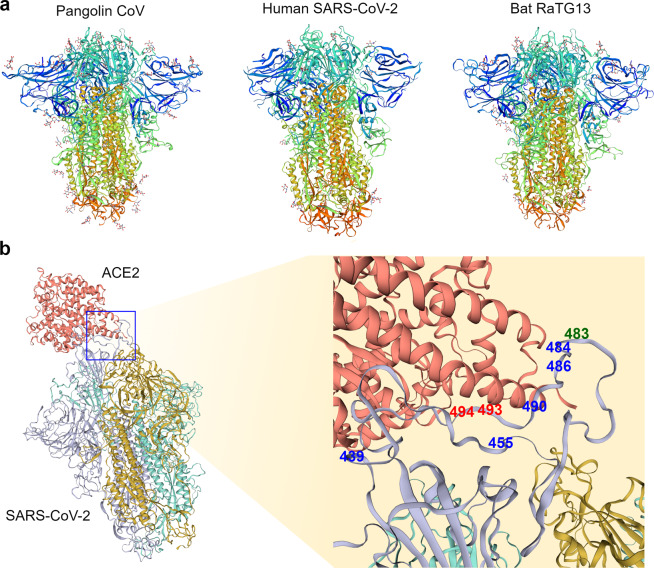
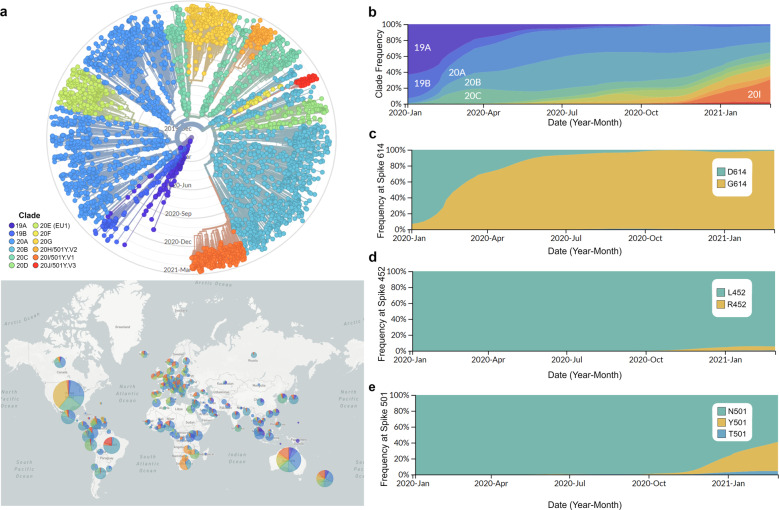
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is responsible for the ongoing global outbreak of a coronavirus disease (herein referred to as COVID-19). Other viruses in the same phylogenetic group have been responsible for previous regional outbreaks, including SARS and MERS. SARS-CoV-2 has a zoonotic origin, similar to the causative viruses of these previous outbreaks. The repetitive introduction of animal viruses into human populations resulting in disease outbreaks suggests that similar future epidemics are inevitable. Therefore, understanding the molecular origin and ongoing evolution of SARS-CoV-2 will provide critical insights for preparing for and preventing future outbreaks. A key feature of SARS-CoV-2 is its propensity for genetic recombination across host species boundaries. Consequently, the genome of SARS-CoV-2 harbors signatures of multiple recombination events, likely encompassing multiple species and broad geographic regions. Other regions of the SARS-CoV-2 genome show the impact of purifying selection. The spike (S) protein of SARS-CoV-2, which enables the virus to enter host cells, exhibits signatures of both purifying selection and ancestral recombination events, leading to an effective S protein capable of infecting human and many other mammalian cells. The global spread and explosive growth of the SARS-CoV-2 population (within human hosts) has contributed additional mutational variability into this genome, increasing opportunities for future recombination.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Woo PCY, et al. Discovery of seven novel mammalian and avian coronaviruses in the genus Deltacoronavirus supports bat coronaviruses as the gene source of Alphacoronavirus, Betacoronavirus and Avian Coronaviruses as the gene source of Gammacoronavirus, Deltacoronavirus. J. Virol. 2012;86:3995. - PMC - PubMed
-
- Jonassen CM, et al. Molecular identification and characterization of novel coronaviruses infecting graylag geese (Anser anser), feral pigeons (Columbia livia) and mallards (Anas platyrhynchos) J. Gen. Virol. 2005;86:1597–1607. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
