

Population-scale tissue transcriptomics maps long non-coding RNAs to complex disease
- PMID: 33864768
- PMCID: PMC8651477
- DOI: 10.1016/j.cell.2021.03.050
Population-scale tissue transcriptomics maps long non-coding RNAs to complex disease
Abstract
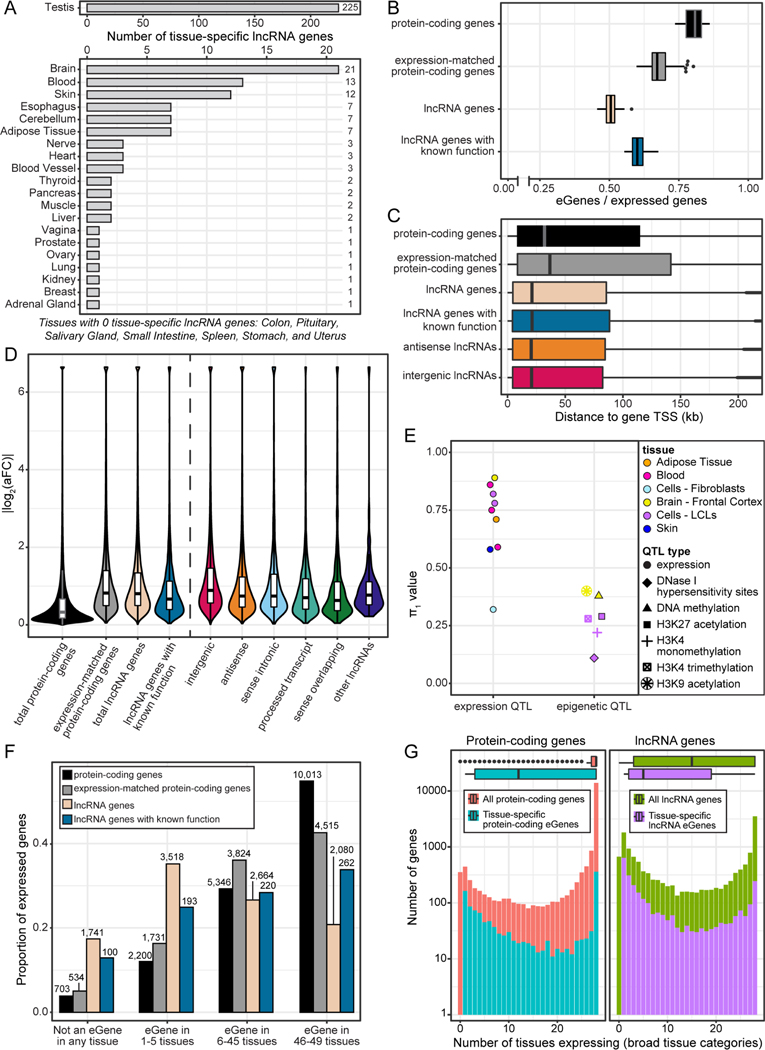
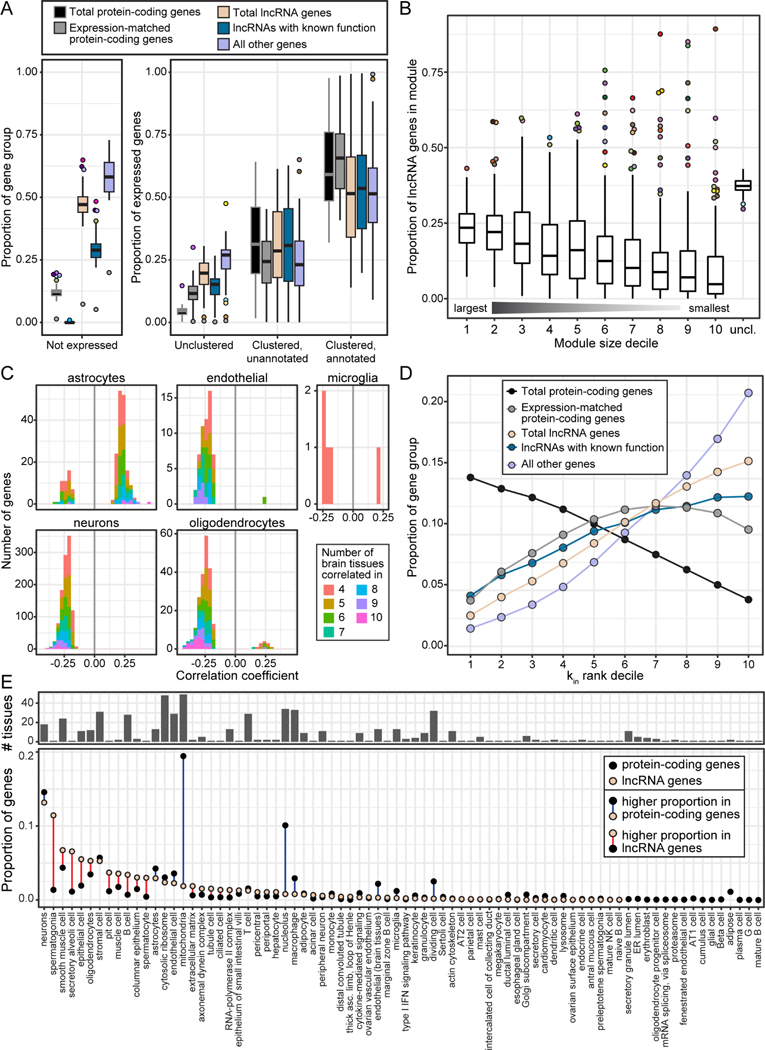
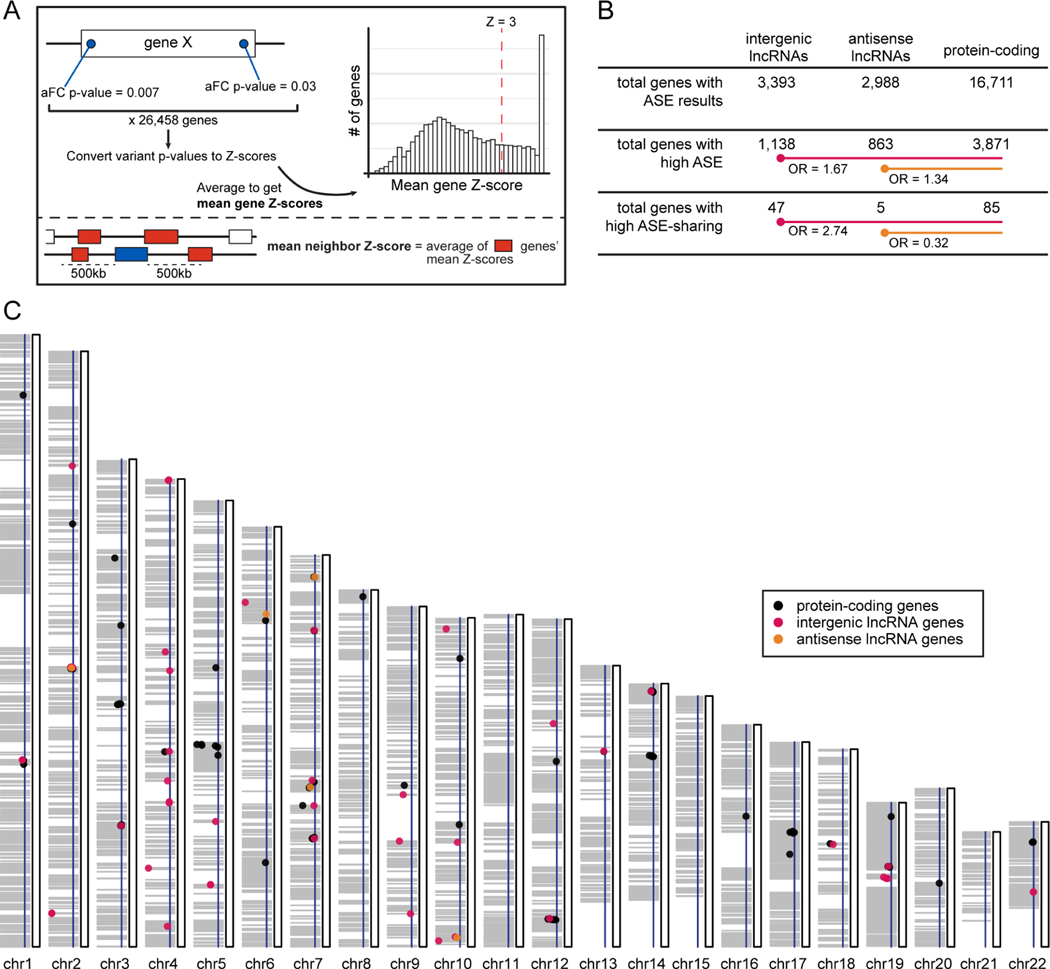
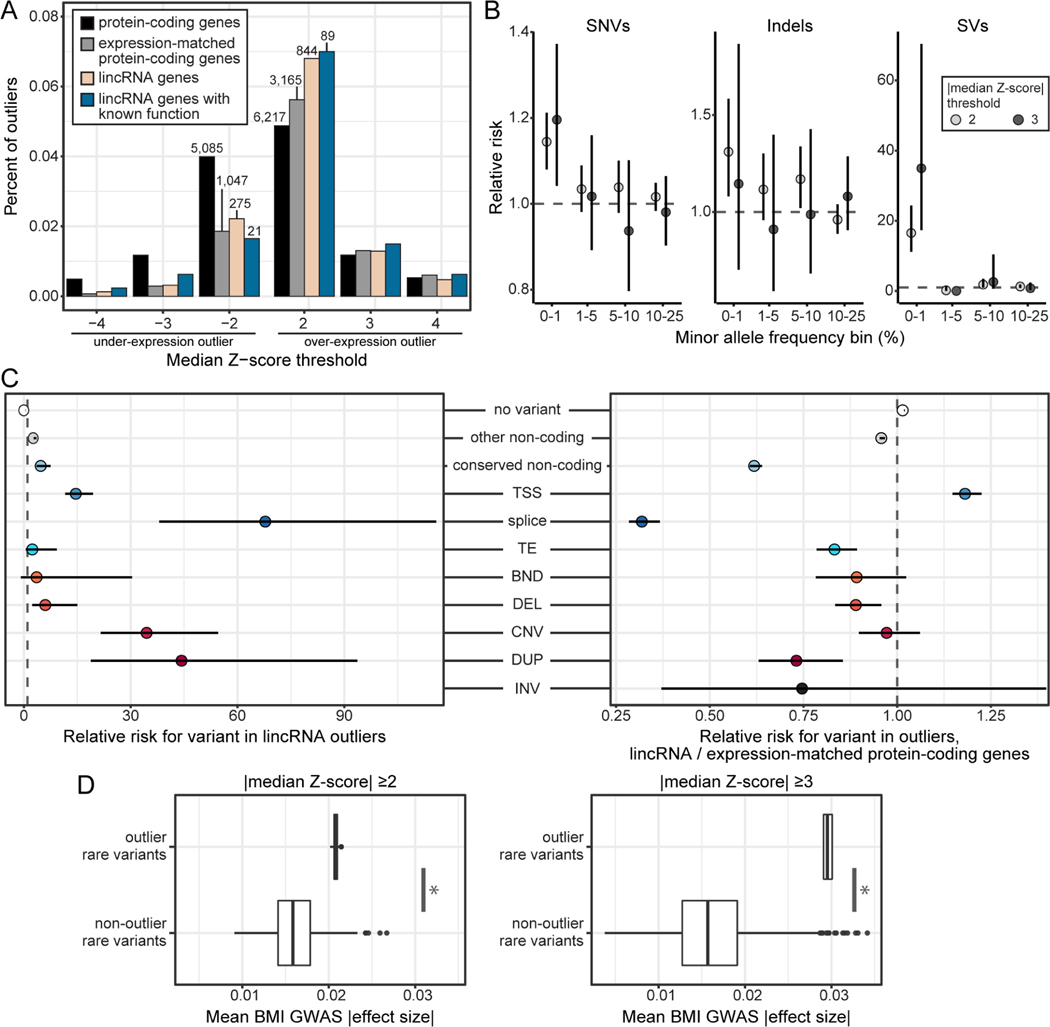
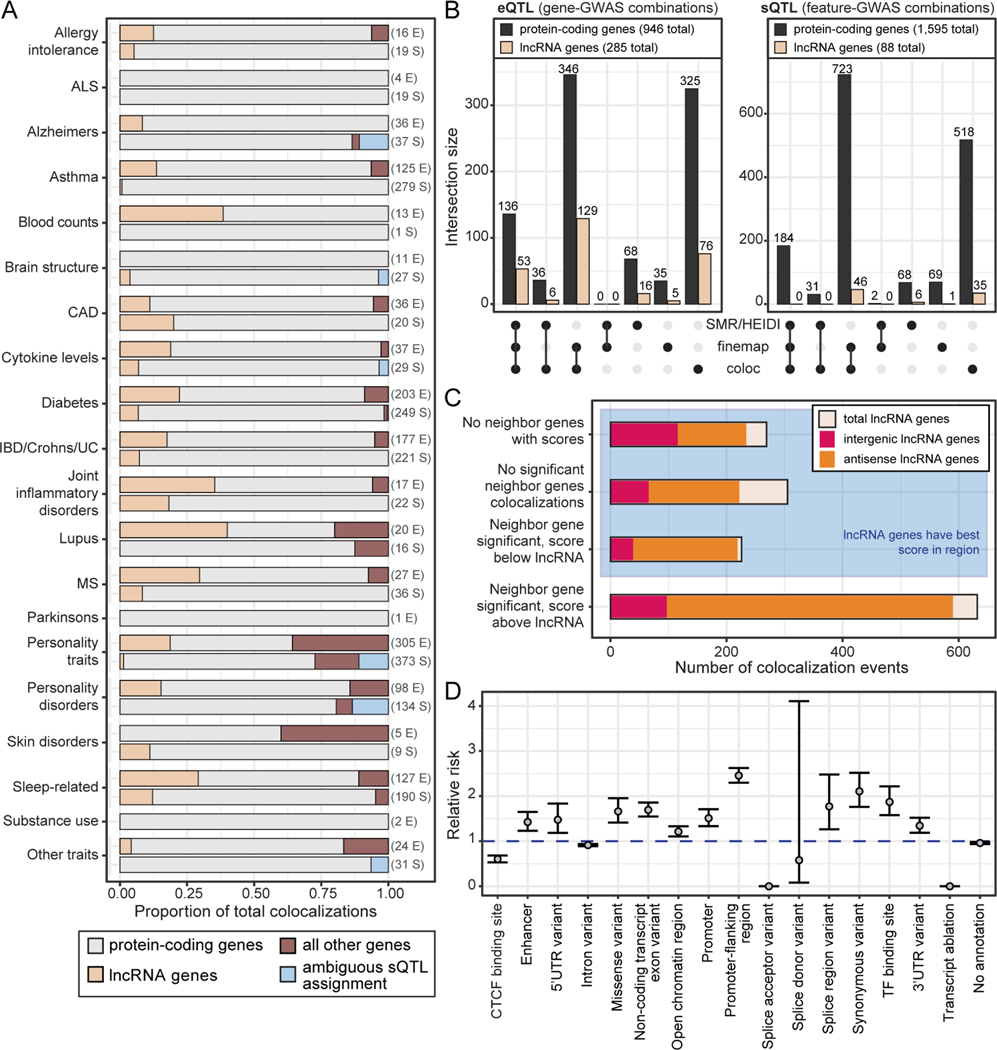
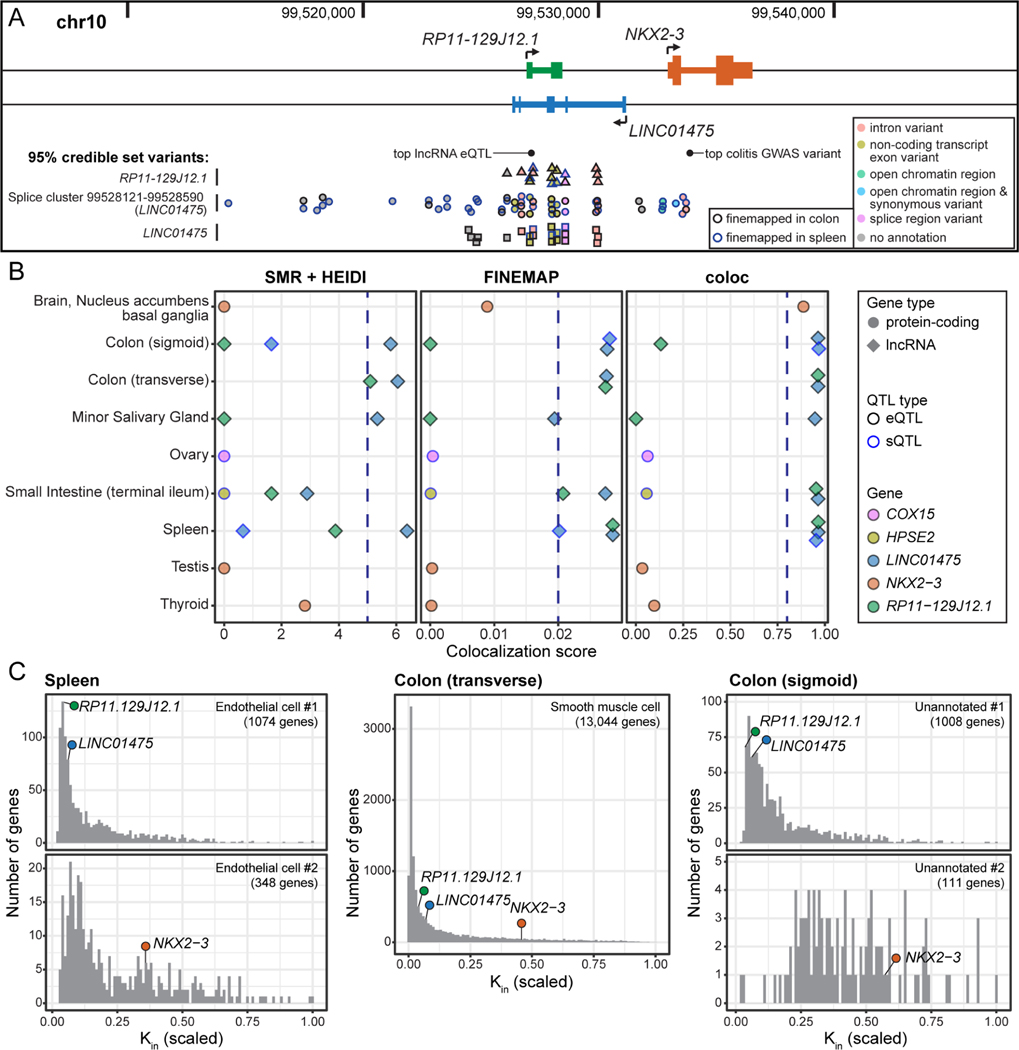
Long non-coding RNA (lncRNA) genes have well-established and important impacts on molecular and cellular functions. However, among the thousands of lncRNA genes, it is still a major challenge to identify the subset with disease or trait relevance. To systematically characterize these lncRNA genes, we used Genotype Tissue Expression (GTEx) project v8 genetic and multi-tissue transcriptomic data to profile the expression, genetic regulation, cellular contexts, and trait associations of 14,100 lncRNA genes across 49 tissues for 101 distinct complex genetic traits. Using these approaches, we identified 1,432 lncRNA gene-trait associations, 800 of which were not explained by stronger effects of neighboring protein-coding genes. This included associations between lncRNA quantitative trait loci and inflammatory bowel disease, type 1 and type 2 diabetes, and coronary artery disease, as well as rare variant associations to body mass index.
Keywords: GTEx; co-expression; colocalization; complex trait; disease; eQTL; expression quantitative trait loci; lncRNA; long non-coding RNA.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests F.A. is an inventor on a patent application related to TensorQTL; S.E.C. is a co-founder and chief technology officer at Variant Bio and owns stock in Variant Bio; T.L. is on the scientific advisory board of Variant Bio, Goldfinch Bio, and GSK and owns stock in Variant Bio; and S.B.M. is on the scientific advisory board of MyOme. All other authors report no competing interests.
Figures
References
-
- Albert FW, and Kruglyak L. (2015). The role of regulatory variation in complex traits and disease. Nature Reviews Genetics 16, 197–212. - PubMed
-
- Allou L, Balzano S, Magg A, Quinodoz M, Royer-Bertrand B, Schöpflin R, Chan W-L, Speck-Martins CE, Carvalho DR, Farage L, et al. (2021). Non-coding deletions identify Maenli lncRNA as a limb-specific En1 regulator. Nature 1–6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL109512/HL/NHLBI NIH HHS/United States
- R01 HG008150/HG/NHGRI NIH HHS/United States
- R01 MH107666/MH/NIMH NIH HHS/United States
- T32 NS105604/NS/NINDS NIH HHS/United States
- R01 MH109905/MH/NIMH NIH HHS/United States
- R01 MH106842/MH/NIMH NIH HHS/United States
- UM1 HG008901/HG/NHGRI NIH HHS/United States
- R01 GM124486/GM/NIGMS NIH HHS/United States
- U01 HG009080/HG/NHGRI NIH HHS/United States
- R01 HL134817/HL/NHLBI NIH HHS/United States
- R01 MH125244/MH/NIMH NIH HHS/United States
- R01 HL135313/HL/NHLBI NIH HHS/United States
- T32 GM007057/GM/NIGMS NIH HHS/United States
- U01 HG009431/HG/NHGRI NIH HHS/United States
- R01 HL139478/HL/NHLBI NIH HHS/United States
- R01 HL142015/HL/NHLBI NIH HHS/United States
- K99 HG009916/HG/NHGRI NIH HHS/United States
- T15 LM007033/LM/NLM NIH HHS/United States
- R33 HL120757/HL/NHLBI NIH HHS/United States
- R01 HL142028/HL/NHLBI NIH HHS/United States
- R01 HG010067/HG/NHGRI NIH HHS/United States
- R01 AG066490/AG/NIA NIH HHS/United States
- R01 GM122924/GM/NIGMS NIH HHS/United States
- U41 HG009494/HG/NHGRI NIH HHS/United States
- R01 MH101822/MH/NIMH NIH HHS/United States
- HHSN268201000029C/HL/NHLBI NIH HHS/United States
- R01 MH101814/MH/NIMH NIH HHS/United States
- P30 DK020595/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
