

Molecular diagnosis of childhood immune dysregulation, polyendocrinopathy, and enteropathy, and implications for clinical management
- PMID: 33864888
- PMCID: PMC8526646
- DOI: 10.1016/j.jaci.2021.04.005
Molecular diagnosis of childhood immune dysregulation, polyendocrinopathy, and enteropathy, and implications for clinical management
Abstract
Background: Most patients with childhood-onset immune dysregulation, polyendocrinopathy, and enteropathy have no genetic diagnosis for their illness. These patients may undergo empirical immunosuppressive treatment with highly variable outcomes.
Objective: We sought to determine the genetic basis of disease in patients referred with Immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like (IPEX-like) disease, but with no mutation in FOXP3; then to assess consequences of genetic diagnoses for clinical management.
Methods: Genomic DNA was sequenced using a panel of 462 genes implicated in inborn errors of immunity. Candidate mutations were characterized by genomic, transcriptional, and (for some) protein analysis.
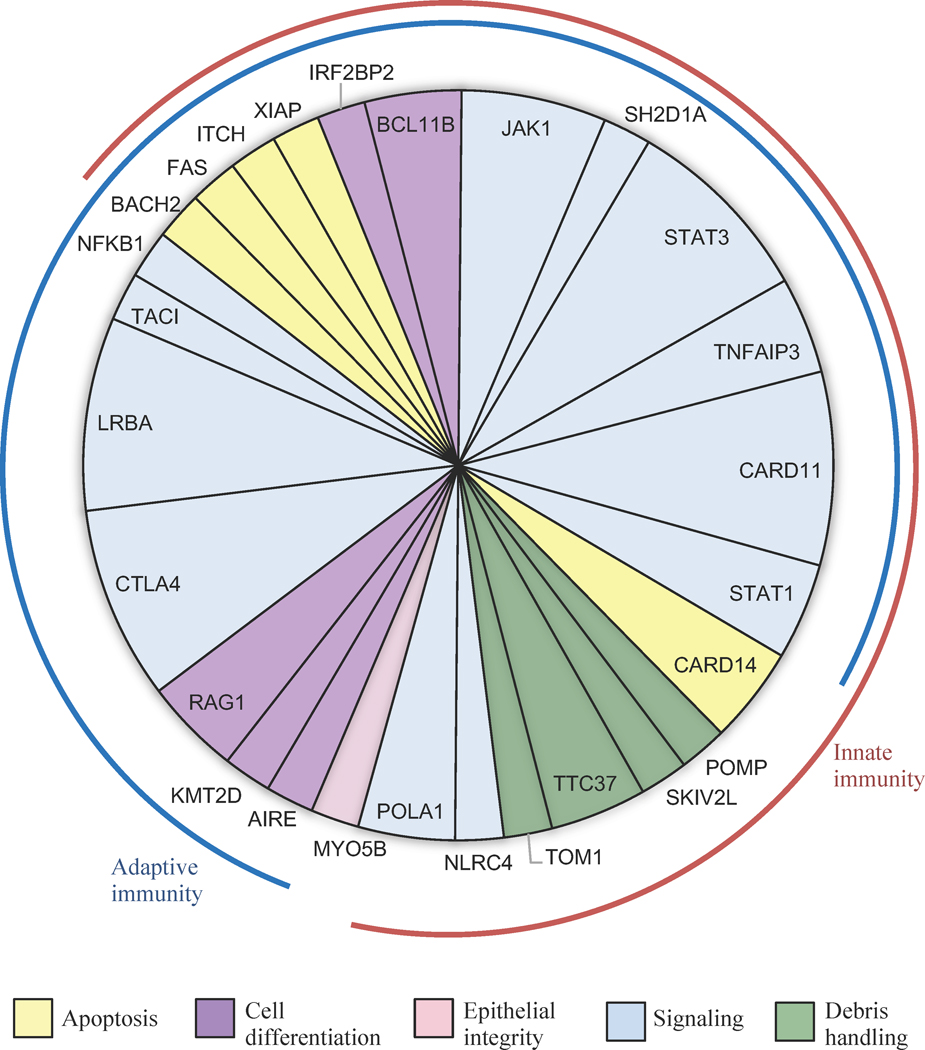
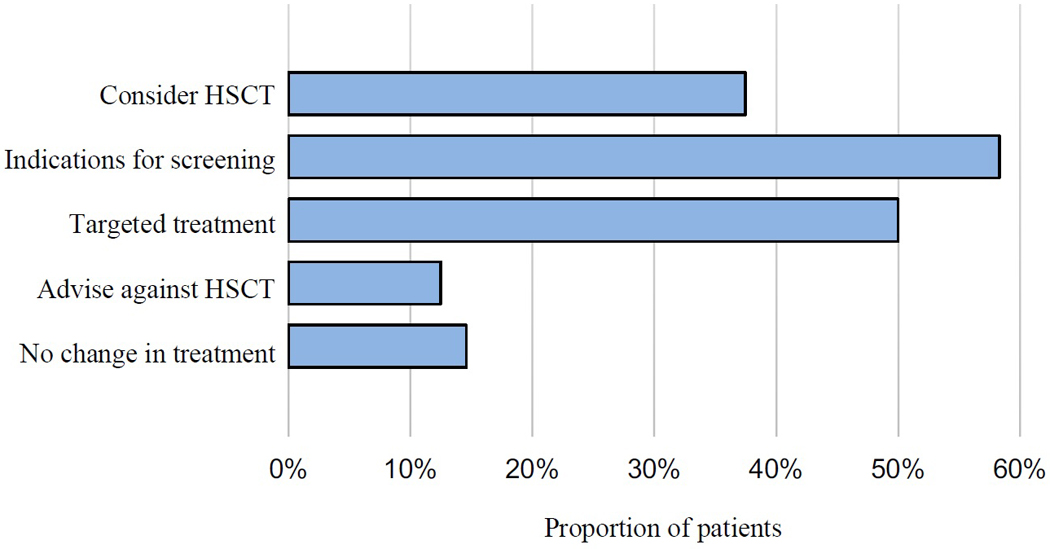
Results: Of 123 patients with FOXP3-negative IPEX-like disease, 48 (39%) carried damaging germline mutations in 1 of the following 27 genes: AIRE, BACH2, BCL11B, CARD11, CARD14, CTLA4, IRF2BP2, ITCH, JAK1, KMT2D, LRBA, MYO5B, NFKB1, NLRC4, POLA1, POMP, RAG1, SH2D1A, SKIV2L, STAT1, STAT3, TNFAIP3, TNFRSF6/FAS, TNRSF13B/TACI, TOM1, TTC37, and XIAP. Many of these genes had not been previously associated with an IPEX-like diagnosis. For 42 of the 48 patients with genetic diagnoses, knowing the critical gene could have altered therapeutic management, including recommendations for targeted treatments and for or against hematopoietic cell transplantation.
Conclusions: Many childhood disorders now bundled as "IPEX-like" disease are caused by individually rare, severe mutations in immune regulation genes. Most genetic diagnoses of these conditions yield clinically actionable findings. Barriers are lack of testing or lack of repeat testing if older technologies failed to provide a diagnosis.
Keywords: Immune dysregulation; autoimmunity; genetics; inborn errors of immunity; molecular diagnosis; pediatric; precision medicine; primary immunodeficiency disorders; sequencing.
Copyright © 2021 American Academy of Allergy, Asthma & Immunology. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Disclosures: Tom Walsh PhD is a consultant for Color Genomics. Troy Torgerson MD PhD is currently employed by the Allen Institute for Immunology, Seattle. The rest of the authors declare that they have no relevant conflicts of interest.
Figures
Similar articles
-
From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation.Ann N Y Acad Sci. 2018 Apr;1417(1):5-22. doi: 10.1111/nyas.13011. Epub 2016 Feb 25. Ann N Y Acad Sci. 2018. PMID: 26918796 Review.
-
IPEX Syndrome: Genetics and Treatment Options.Genes (Basel). 2021 Feb 24;12(3):323. doi: 10.3390/genes12030323. Genes (Basel). 2021. PMID: 33668198 Free PMC article. Review.
-
Persistent Enteropathy in a Toddler with a Novel FOXP3 Mutation and Normal FOXP3 Protein Expression.J Pediatr. 2017 Jul;186:183-185. doi: 10.1016/j.jpeds.2017.03.051. Epub 2017 Apr 27. J Pediatr. 2017. PMID: 28457527
-
[A case of immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome with very early onset inflammatory bowel disease-like changes].Zhonghua Er Ke Za Zhi. 2019 Jul 2;57(7):559-561. doi: 10.3760/cma.j.issn.0578-1310.2019.07.013. Zhonghua Er Ke Za Zhi. 2019. PMID: 31269558 Chinese.
-
A challenging undertaking: Stem cell transplantation for immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome.J Allergy Clin Immunol. 2016 Mar;137(3):953-5.e4. doi: 10.1016/j.jaci.2015.09.030. Epub 2015 Nov 11. J Allergy Clin Immunol. 2016. PMID: 26559324 No abstract available.
Cited by
-
Novel frameshift variants expand the map of the genetic defects in IRF2BP2.Front Immunol. 2023 Oct 9;14:1279171. doi: 10.3389/fimmu.2023.1279171. eCollection 2023. Front Immunol. 2023. PMID: 37876937 Free PMC article.
-
Understanding inborn errors of immunity: A lens into the pathophysiology of monogenic inflammatory bowel disease.Front Immunol. 2022 Sep 29;13:1026511. doi: 10.3389/fimmu.2022.1026511. eCollection 2022. Front Immunol. 2022. PMID: 36248828 Free PMC article. Review.
-
The role of the CBM complex in allergic inflammation and disease.J Allergy Clin Immunol. 2022 Nov;150(5):1011-1030. doi: 10.1016/j.jaci.2022.06.023. Epub 2022 Aug 16. J Allergy Clin Immunol. 2022. PMID: 35981904 Free PMC article. Review.
-
The genetics of monogenic intestinal epithelial disorders.Hum Genet. 2023 May;142(5):613-654. doi: 10.1007/s00439-022-02501-5. Epub 2022 Nov 23. Hum Genet. 2023. PMID: 36422736 Free PMC article. Review.
-
IRF2BP2 binds to a conserved RxSVI motif of protein partners and regulates megakaryocytic differentiation.Nat Commun. 2024 Nov 30;15(1):10425. doi: 10.1038/s41467-024-54889-5. Nat Commun. 2024. PMID: 39616187 Free PMC article.
References
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
