

Type I Interferonopathies in Children: An Overview
- PMID: 33869112
- PMCID: PMC8044321
- DOI: 10.3389/fped.2021.631329
Type I Interferonopathies in Children: An Overview
Abstract
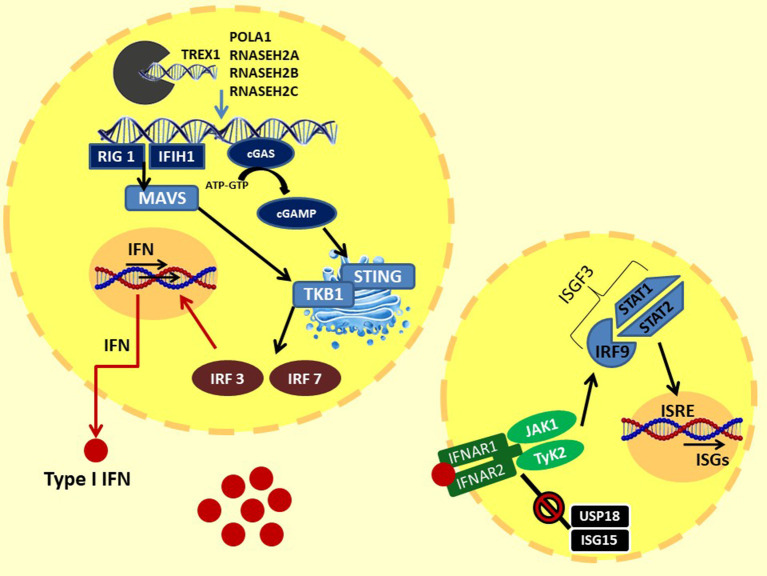
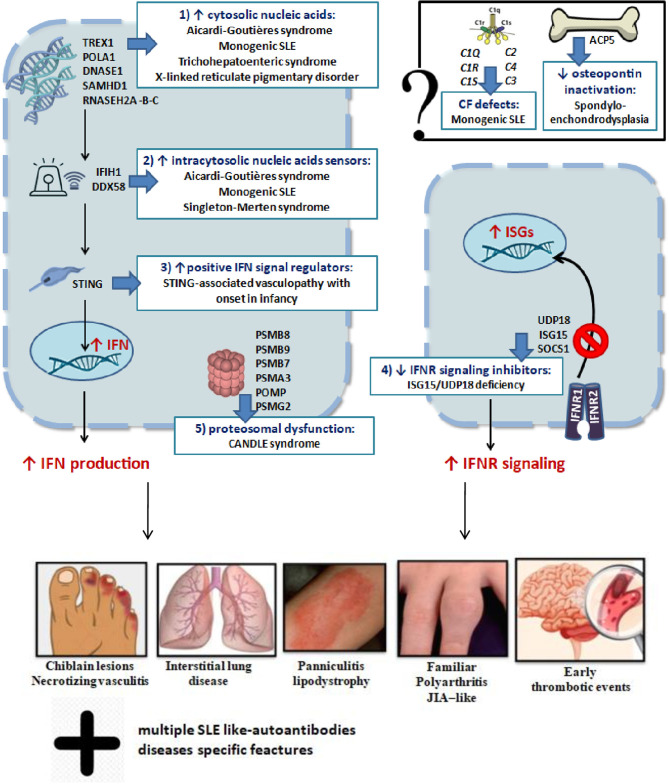
Notable advances in gene sequencing methods in recent years have permitted enormous progress in the phenotypic and genotypic characterization of autoinflammatory syndromes. Interferonopathies are a recent group of inherited autoinflammatory diseases, characterized by a dysregulation of the interferon pathway, leading to constitutive upregulation of its activation mechanisms or downregulation of negative regulatory systems. They are clinically heterogeneous, but some peculiar clinical features may lead to suspicion: a familial "idiopathic" juvenile arthritis resistant to conventional treatments, an early necrotizing vasculitis, a non-infectious interstitial lung disease, and a panniculitis associated or not with a lipodystrophy may represent the "interferon alarm bells." The awareness of this group of diseases represents a challenge for pediatricians because, despite being rare, a differential diagnosis with the most common childhood rheumatological and immunological disorders is mandatory. Furthermore, the characterization of interferonopathy molecular pathogenetic mechanisms is allowing important steps forward in other immune dysregulation diseases, such as systemic lupus erythematosus and inflammatory myositis, implementing the opportunity of a more effective target therapy.
Keywords: Aicardi-Goutières syndrome; Janus kinase inhibitors; autoinflammatory disease; innate immunity; interferon; type I interferon (IFN) signaling.
Copyright © 2021 d'Angelo, Di Filippo, Breda and Chiarelli.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources