

Overexpression of SERCA2a Alleviates Cardiac Microvascular Ischemic Injury by Suppressing Mfn2-Mediated ER/Mitochondrial Calcium Tethering
- PMID: 33869181
- PMCID: PMC8047138
- DOI: 10.3389/fcell.2021.636553
Overexpression of SERCA2a Alleviates Cardiac Microvascular Ischemic Injury by Suppressing Mfn2-Mediated ER/Mitochondrial Calcium Tethering
Retraction in
-
Retraction: Overexpression of SERCA2a alleviates cardiac microvascular ischemic injury by suppressing Mfn2-mediated ER/mitochondrial calcium tethering.Front Cell Dev Biol. 2022 Aug 4;10:1006540. doi: 10.3389/fcell.2022.1006540. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 35990600 Free PMC article.
Abstract
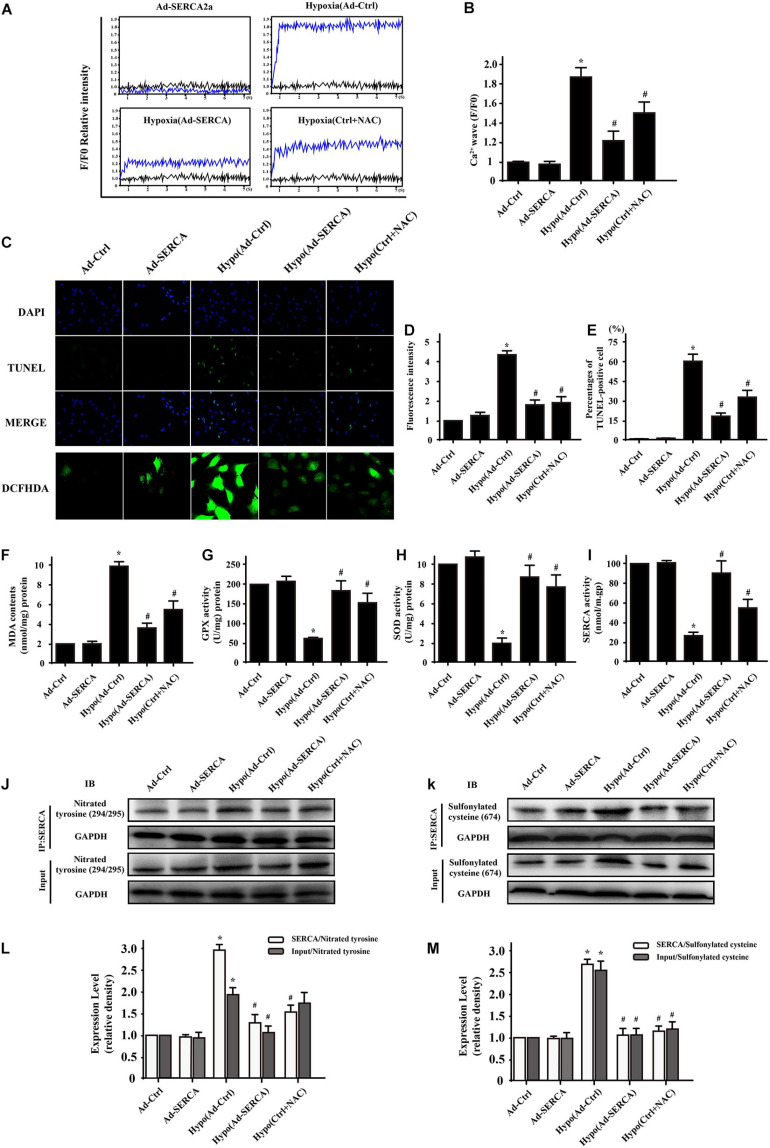
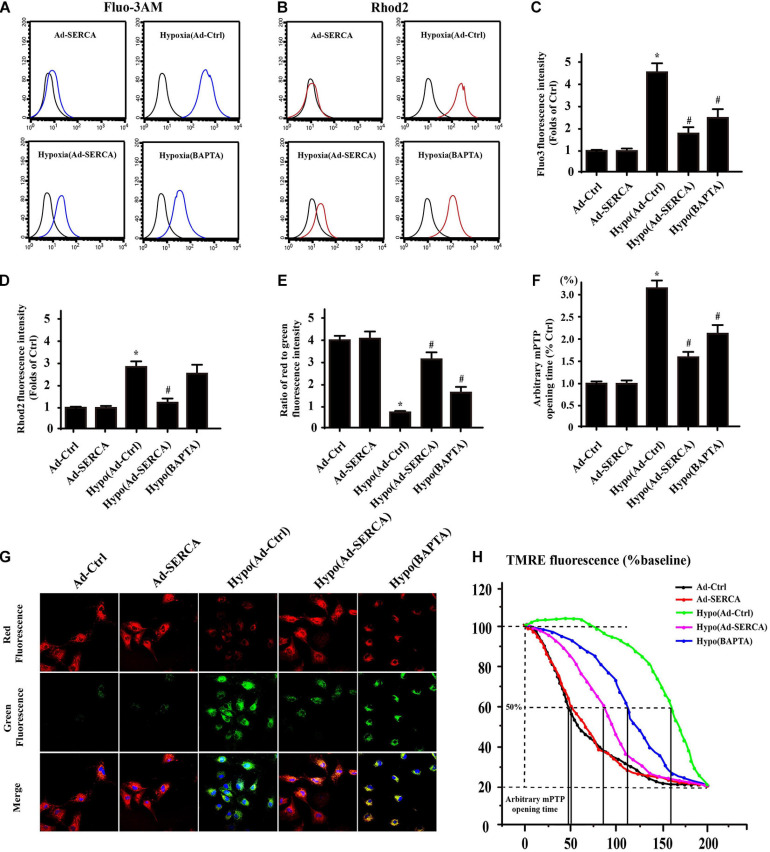
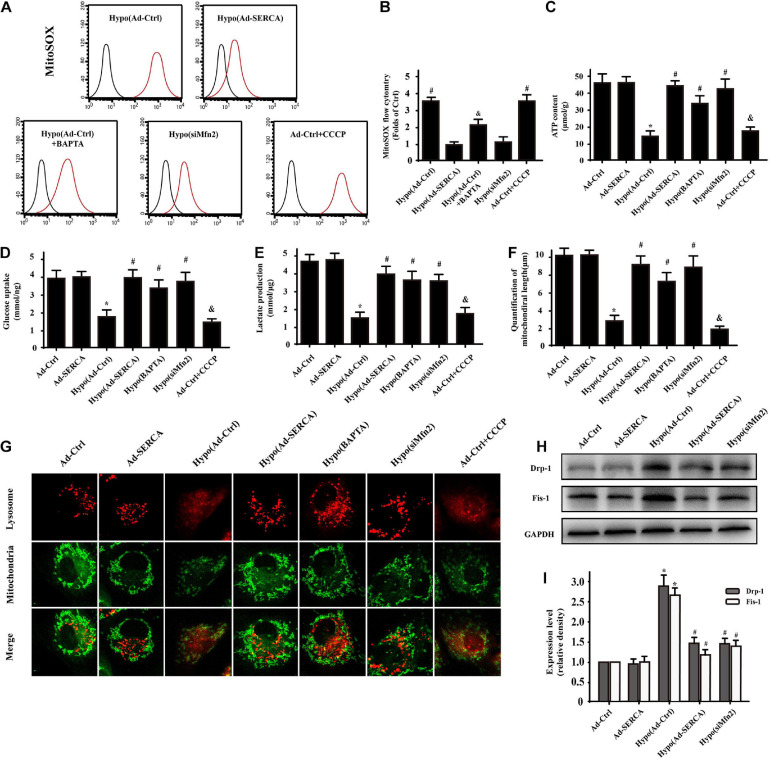
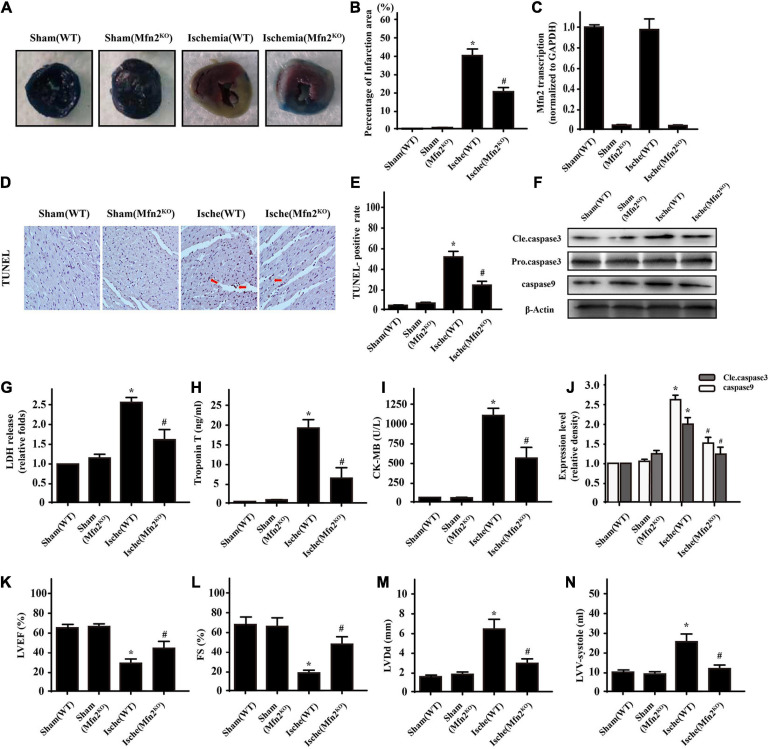
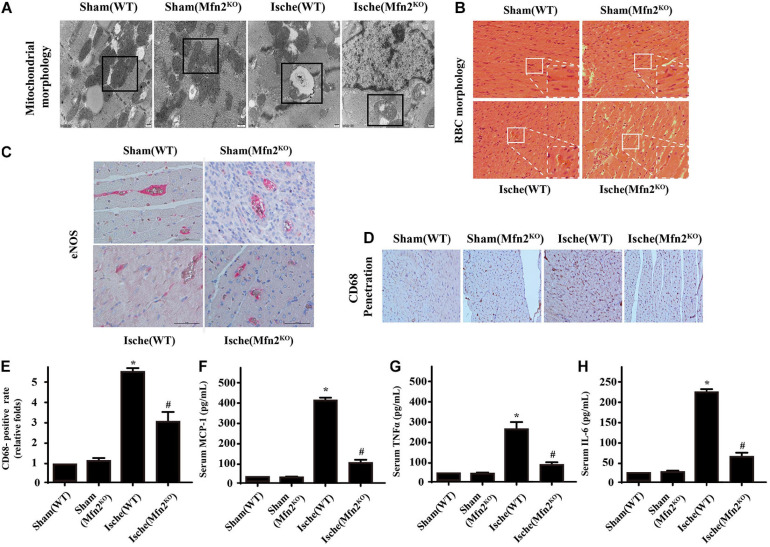
Our previous research has shown that type-2a Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA2a) undergoes posttranscriptional oxidative modifications in cardiac microvascular endothelial cells (CMECs) in the context of excessive cardiac oxidative injury. However, whether SERCA2a inactivity induces cytosolic Ca2+ imbalance in mitochondrial homeostasis is far from clear. Mitofusin2 (Mfn2) is well known as an important protein involved in endoplasmic reticulum (ER)/mitochondrial Ca2+ tethering and the regulation of mitochondrial quality. Therefore, the aim of our study was to elucidate the specific mechanism of SERCA2a-mediated Ca2+ overload in the mitochondria via Mfn2 tethering and the survival rate of the heart under conditions of cardiac microvascular ischemic injury. In vitro, CMECs extracted from mice were subjected to 6 h of hypoxic injury to mimic ischemic heart injury. C57-WT and Mfn2KO mice were subjected to a 1 h ischemia procedure via ligation of the left anterior descending branch to establish an in vivo cardiac ischemic injury model. TTC staining, immunohistochemistry and echocardiography were used to assess the myocardial infarct size, microvascular damage, and heart function. In vitro, ischemic injury induced irreversible oxidative modification of SERCA2a, including sulfonylation at cysteine 674 and nitration at tyrosine 294/295, and inactivation of SERCA2a, which initiated calcium overload. In addition, ischemic injury-triggered [Ca2+]c overload and subsequent [Ca2+]m overload led to mPTP opening and ΔΨm dissipation compared with the control. Furthermore, ablation of Mfn2 alleviated SERCA2a-induced mitochondrial calcium overload and subsequent mito-apoptosis in the context of CMEC hypoxic injury. In vivo, compared with that in wild-type mice, the myocardial infarct size in Mfn2KO mice was significantly decreased. In addition, the findings revealed that Mfn2KO mice had better heart contractile function, decreased myocardial infarction indicators, and improved mitochondrial morphology. Taken together, the results of our study suggested that SERCA2a-dependent [Ca2+]c overload led to mitochondrial dysfunction and activation of Mfn2-mediated [Ca2+]m overload. Overexpression of SERCA2a or ablation of Mfn2 expression mitigated mitochondrial morphological and functional damage by modifying the SERCA2a/Ca2+-Mfn2 pathway. Overall, these pathways are promising therapeutic targets for acute cardiac microvascular ischemic injury.
Keywords: CMEC; Mfn2; SERCA2a; hypoxia; ischemia injury; mitochondria.
Copyright © 2021 Tian and Zhang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures


Similar articles
-
Protective effect of HINT2 on mitochondrial function via repressing MCU complex activation attenuates cardiac microvascular ischemia-reperfusion injury.Basic Res Cardiol. 2021 Dec 16;116(1):65. doi: 10.1007/s00395-021-00905-4. Basic Res Cardiol. 2021. PMID: 34914018 Free PMC article.
-
Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction.Cell Death Dis. 2016 May 26;7(5):e2238. doi: 10.1038/cddis.2016.139. Cell Death Dis. 2016. PMID: 27228353 Free PMC article.
-
Liraglutide protects cardiac microvascular endothelial cells against hypoxia/reoxygenation injury through the suppression of the SR-Ca(2+)-XO-ROS axis via activation of the GLP-1R/PI3K/Akt/survivin pathways.Free Radic Biol Med. 2016 Jun;95:278-92. doi: 10.1016/j.freeradbiomed.2016.03.035. Epub 2016 Mar 31. Free Radic Biol Med. 2016. PMID: 27038735
-
Cardiac SERCA2A/B: therapeutic targets for heart failure.Eur J Pharmacol. 2014 Feb 5;724:1-8. doi: 10.1016/j.ejphar.2013.12.018. Epub 2013 Dec 18. Eur J Pharmacol. 2014. PMID: 24361307 Review.
-
Decoding interaction between mitochondria and endoplasmic reticulum in ischemic myocardial injury: targeting natural medicines.Front Pharmacol. 2025 Feb 28;16:1536773. doi: 10.3389/fphar.2025.1536773. eCollection 2025. Front Pharmacol. 2025. PMID: 40093324 Free PMC article. Review.
Cited by
-
Parkin Insufficiency Accentuates High-Fat Diet-Induced Cardiac Remodeling and Contractile Dysfunction Through VDAC1-Mediated Mitochondrial Ca2+ Overload.JACC Basic Transl Sci. 2022 Aug 22;7(8):779-796. doi: 10.1016/j.jacbts.2022.03.007. eCollection 2022 Aug. JACC Basic Transl Sci. 2022. PMID: 36061337 Free PMC article.
-
Mitochondrial quality control in cardiac ischemia/reperfusion injury: new insights into mechanisms and implications.Cell Biol Toxicol. 2023 Feb;39(1):33-51. doi: 10.1007/s10565-022-09716-2. Epub 2022 Aug 11. Cell Biol Toxicol. 2023. PMID: 35951200 Review.
-
Autophagy protects mitochondrial health in heart failure.Heart Fail Rev. 2024 Jan;29(1):113-123. doi: 10.1007/s10741-023-10354-x. Epub 2023 Oct 12. Heart Fail Rev. 2024. PMID: 37823952 Review.
-
Advances in the mechanism and treatment of mitochondrial quality control involved in myocardial infarction.J Cell Mol Med. 2021 Aug;25(15):7110-7121. doi: 10.1111/jcmm.16744. Epub 2021 Jun 23. J Cell Mol Med. 2021. PMID: 34160885 Free PMC article. Review.
-
Protective effect of HINT2 on mitochondrial function via repressing MCU complex activation attenuates cardiac microvascular ischemia-reperfusion injury.Basic Res Cardiol. 2021 Dec 16;116(1):65. doi: 10.1007/s00395-021-00905-4. Basic Res Cardiol. 2021. PMID: 34914018 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous