

A single-cell genomics pipeline for environmental microbial eukaryotes
- PMID: 33870123
- PMCID: PMC8042348
- DOI: 10.1016/j.isci.2021.102290
A single-cell genomics pipeline for environmental microbial eukaryotes
Abstract
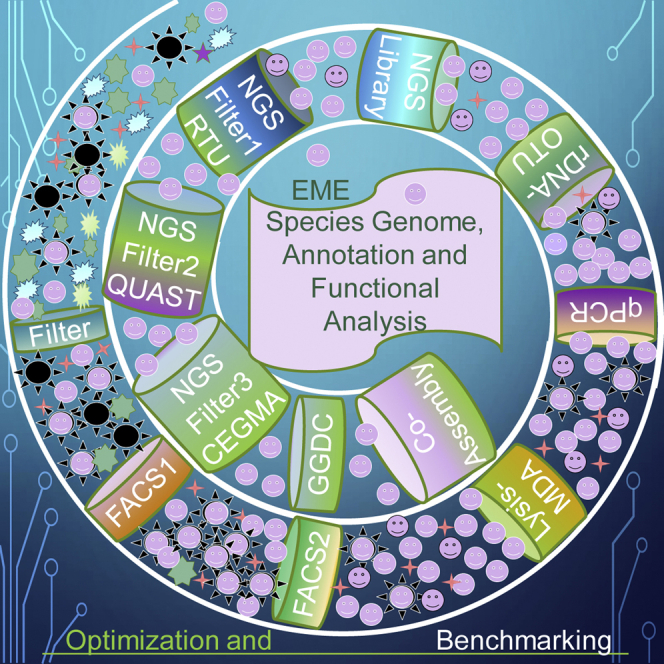
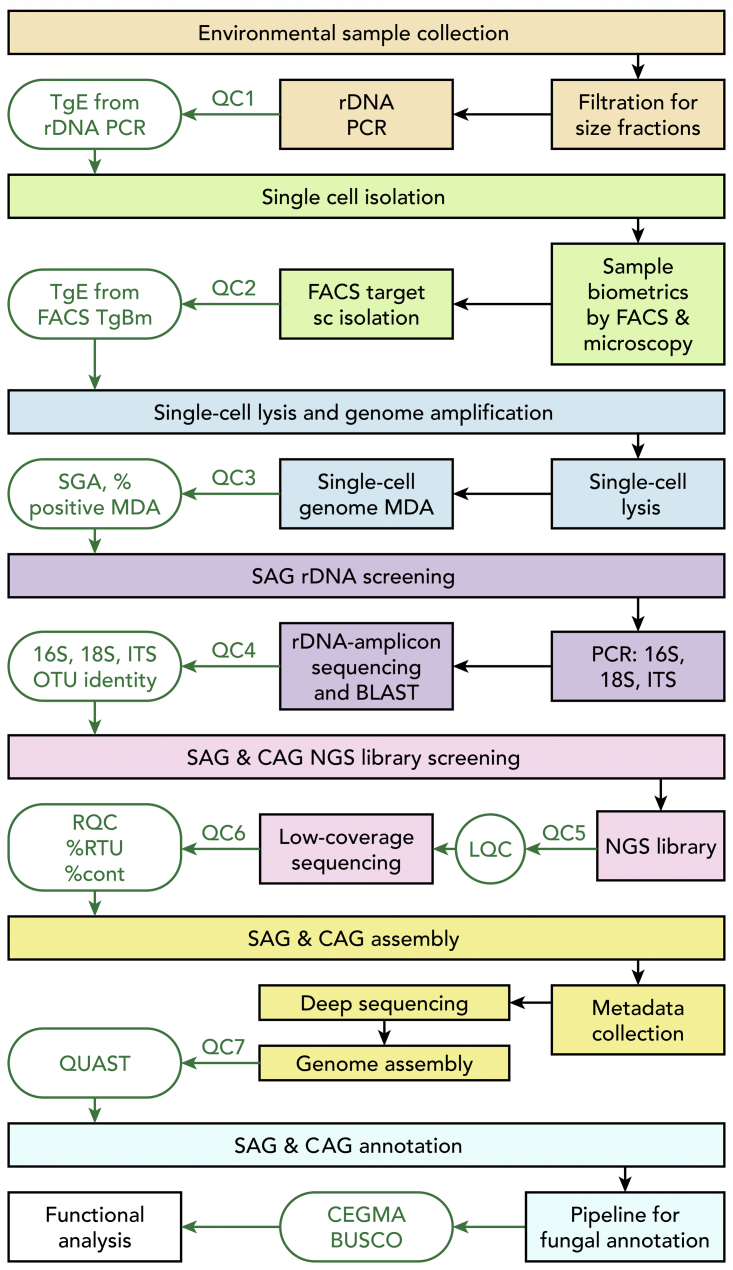
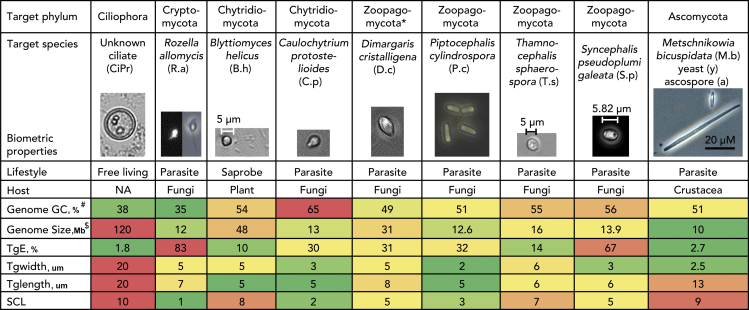
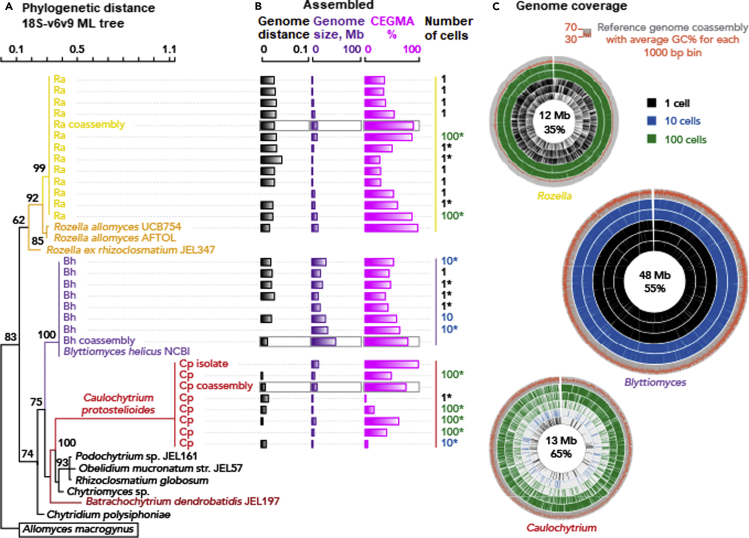
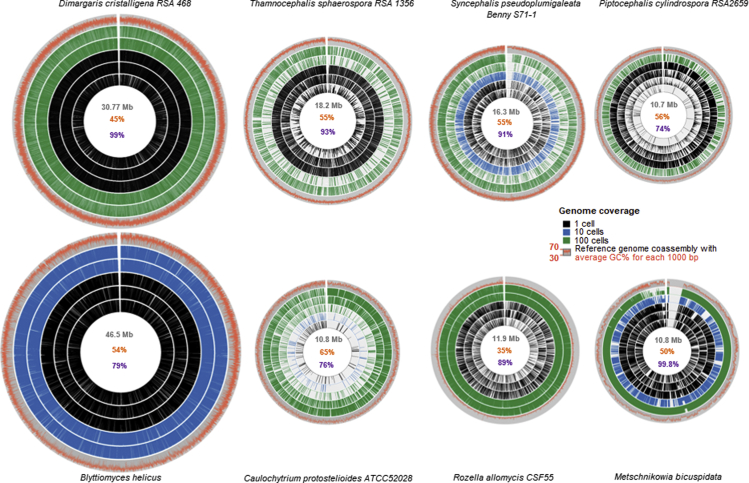
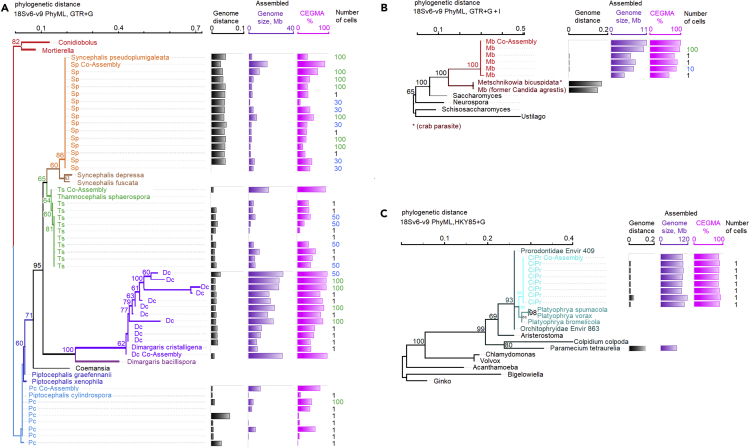
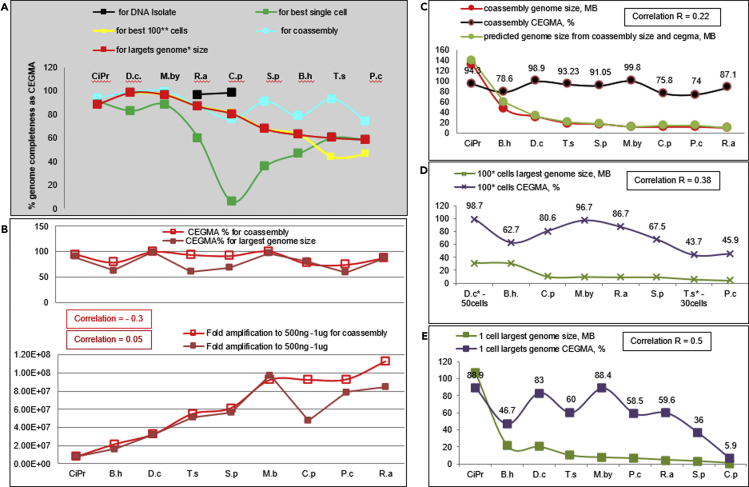
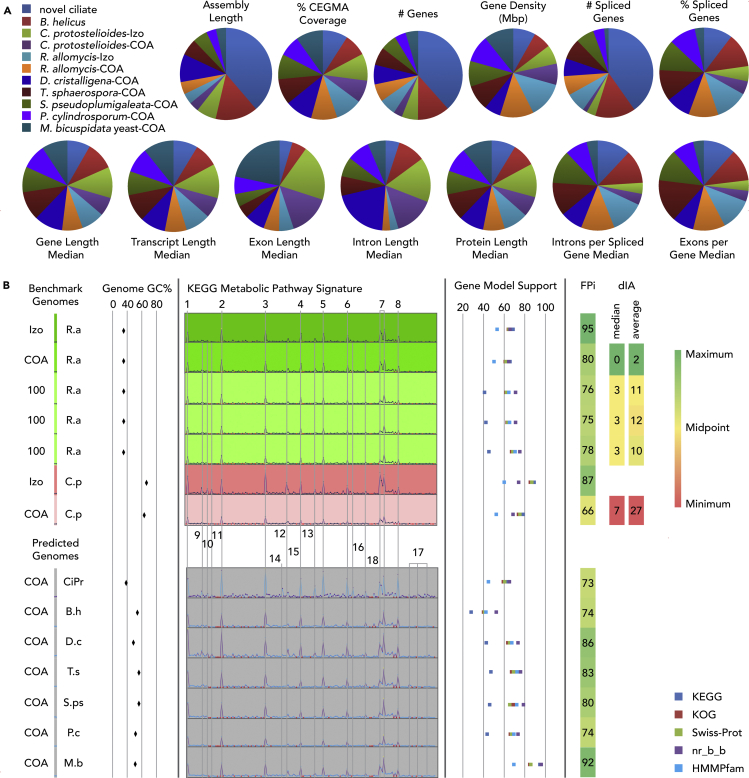
Single-cell sequencing of environmental microorganisms is an essential component of the microbial ecology toolkit. However, large-scale targeted single-cell sequencing for the whole-genome recovery of uncultivated eukaryotes is lagging. The key challenges are low abundance in environmental communities, large complex genomes, and cell walls that are difficult to break. We describe a pipeline composed of state-of-the art single-cell genomics tools and protocols optimized for poorly studied and uncultivated eukaryotic microorganisms that are found at low abundance. This pipeline consists of seven distinct steps, beginning with sample collection and ending with genome annotation, each equipped with quality review steps to ensure high genome quality at low cost. We tested and evaluated each step on environmental samples and cultures of early-diverging lineages of fungi and Chromista/SAR. We show that genomes produced using this pipeline are almost as good as complete reference genomes for functional and comparative genomics for environmental microbial eukaryotes.
Keywords: Genomics; Geomicrobiology; Microbiology.
Figures

References
-
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. - PubMed
-
- Arriola E., Lambros M.B., Jones C., Dexter T., Mackay A., Tan D.S., Tamber N., Fenwick K., Ashworth A., Dowsett M. Evaluation of Phi29-based whole-genome amplification for microarray-based comparative genomic hybridisation. Lab. Invest. 2007;87:75–83. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
