

The Metabolic Landscape of RAS-Driven Cancers from biology to therapy
- PMID: 33870211
- PMCID: PMC8045781
- DOI: 10.1038/s43018-021-00184-x
The Metabolic Landscape of RAS-Driven Cancers from biology to therapy
Abstract
Our understanding of how the RAS protein family, and in particular mutant KRAS promote metabolic dysregulation in cancer cells has advanced significantly over the last decade. In this Review, we discuss the metabolic reprogramming mediated by oncogenic RAS in cancer, and elucidating the underlying mechanisms could translate to novel therapeutic opportunities to target metabolic vulnerabilities in RAS-driven cancers.
Keywords: KRAS; autophagy; cancer therapeutics; chemoresistance; ferroptosis; glutaminolysis; glycolysis; macropinocytosis; metabolism.
Conflict of interest statement
Competing Interests The authors are aware of no direct conflicts with the topic of the paper; however, M.G.V.H. is a Scientic Advisory Board member for Agios Pharmaceuticals, Aeglea Biotherapetics, iTeos Therapeutics, Faeth Therapeutics, and Auron Therapeutics. F.M. is a consultant for the following companies: Amgen; Daiichi Ltd.; Ideaya Biosciences; Kura Oncology; Leidos Biomedical Research, Inc.; PellePharm; Pfizer Inc.; PMV Pharma and Quanta Therapeutics. F.M. is a consultant and co-founder for the following companies (with ownership interest including stock options): BridgeBio; DNAtrix Inc.; Olema Pharmaceuticals, Inc.; and Quartz. F.M. is the scientific director of the National Cancer Institute (NCI) RAS Initiative at the Frederick National Laboratory for Cancer Research/Leidos Biomedical Research, Inc. None of these affiliations represents a conflict of interest with respect to this manuscript.
Figures
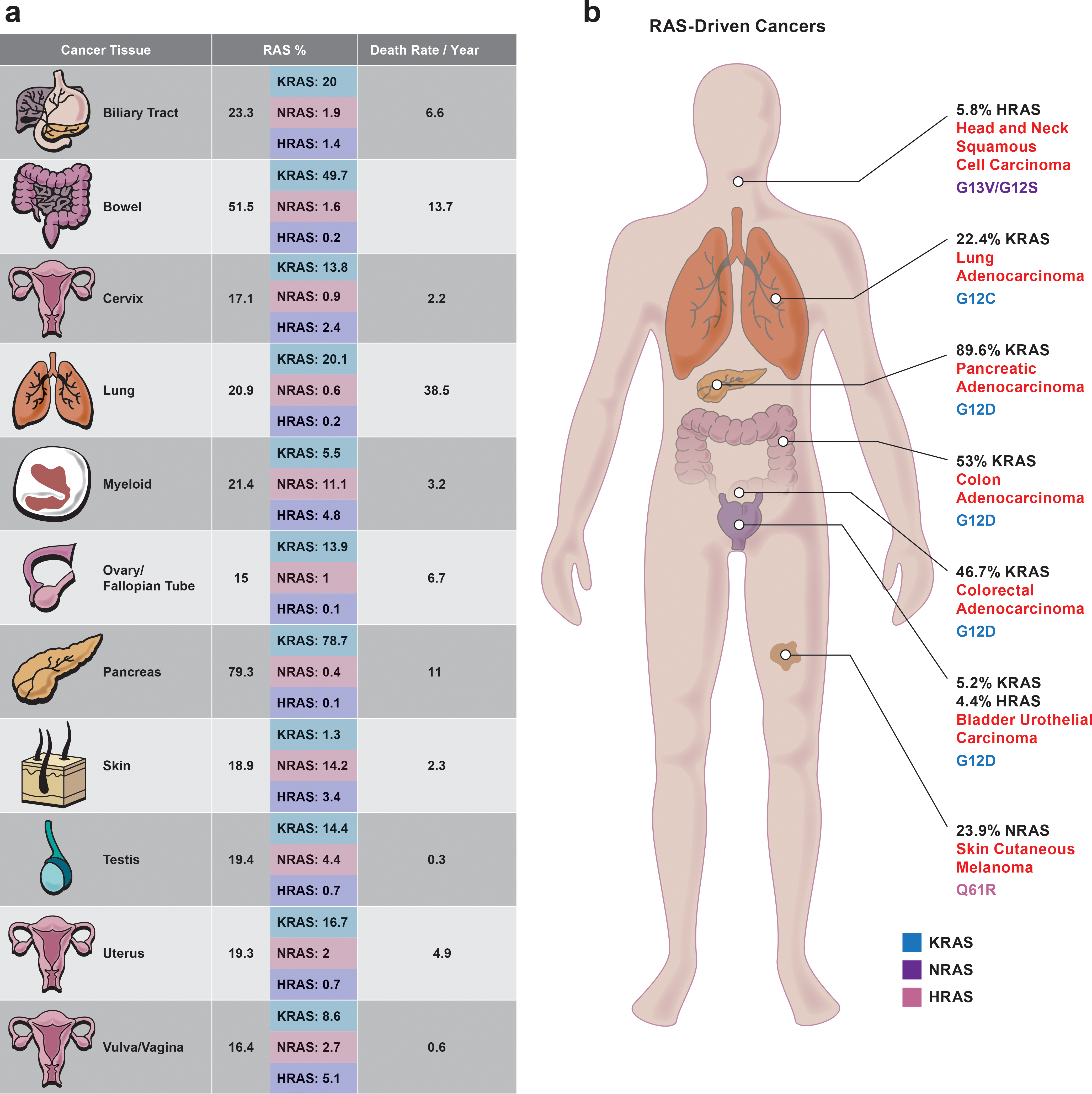
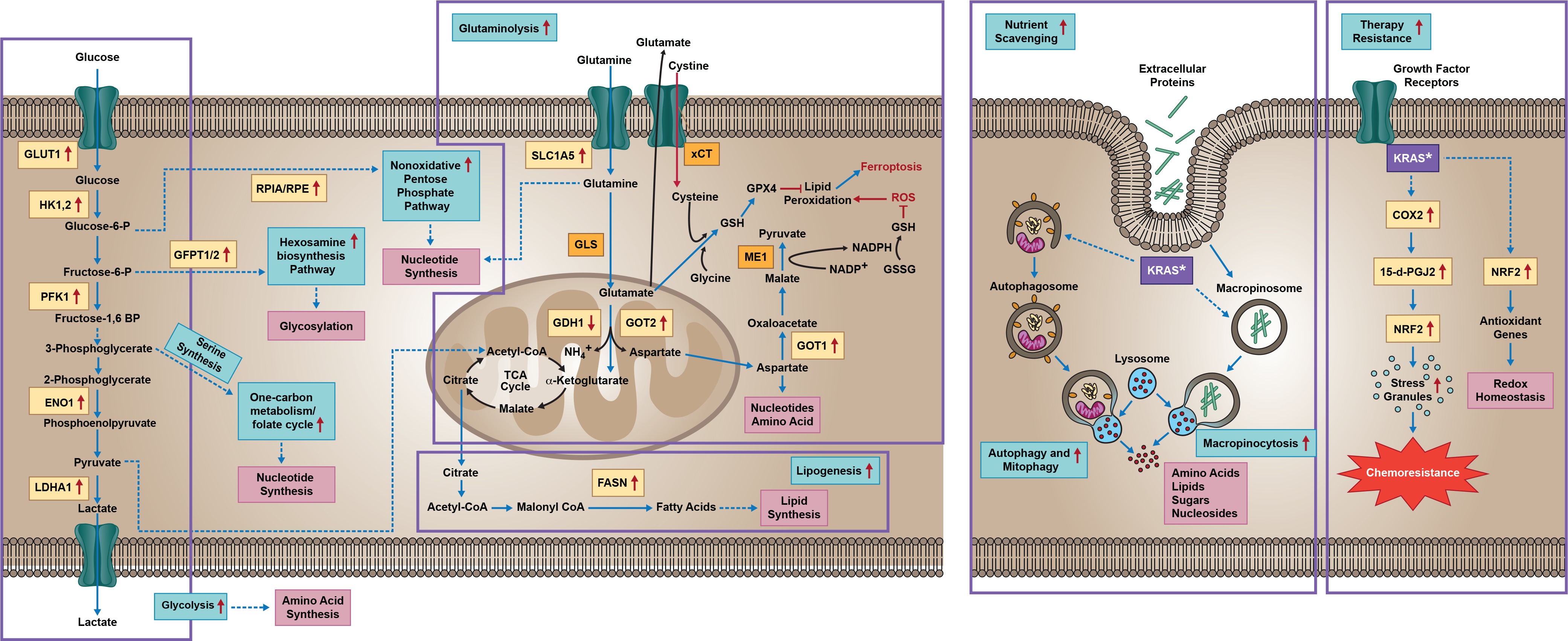
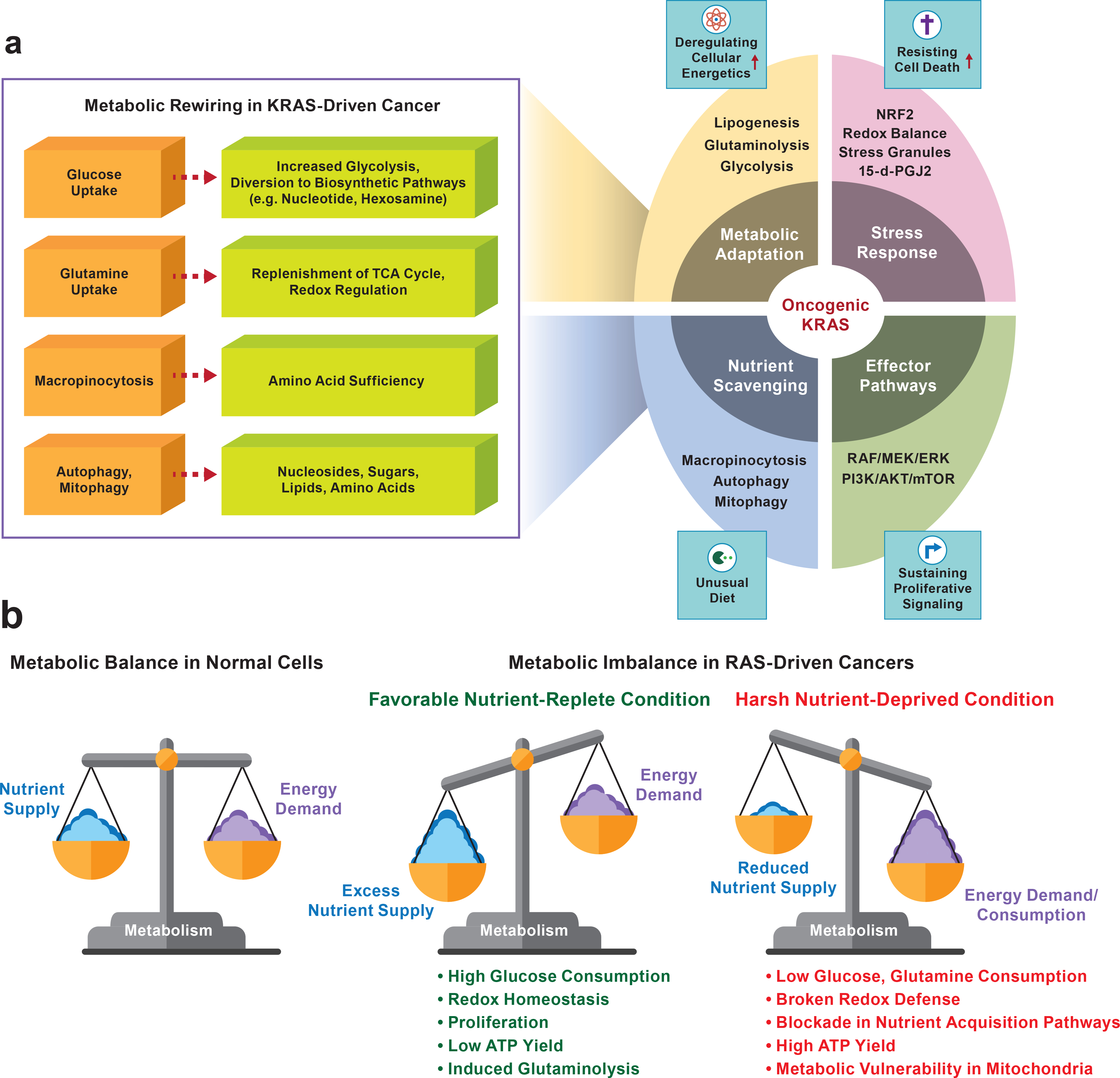
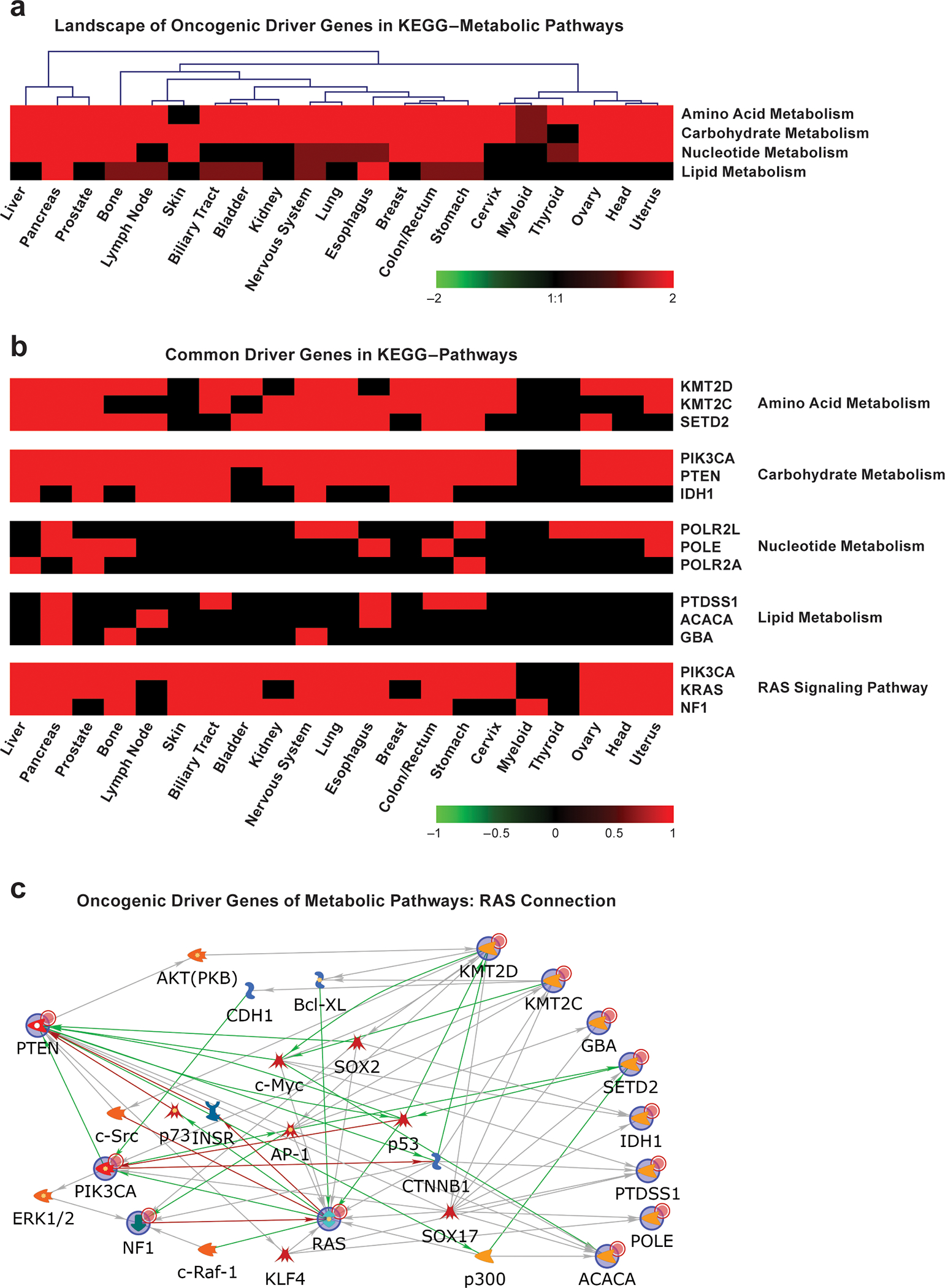
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
