

Inferring Adaptive Codon Preference to Understand Sources of Selection Shaping Codon Usage Bias
- PMID: 33871580
- PMCID: PMC8321536
- DOI: 10.1093/molbev/msab099
Inferring Adaptive Codon Preference to Understand Sources of Selection Shaping Codon Usage Bias
Abstract
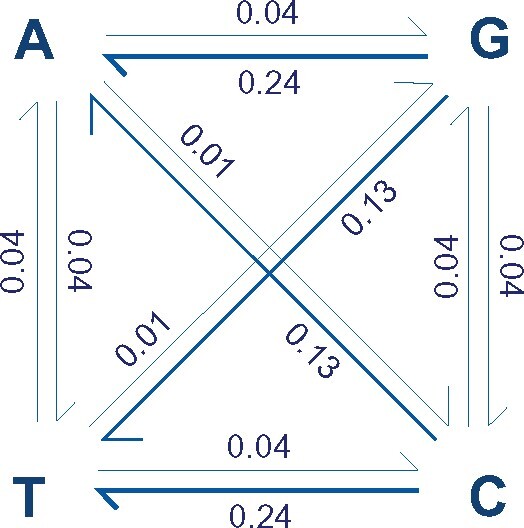
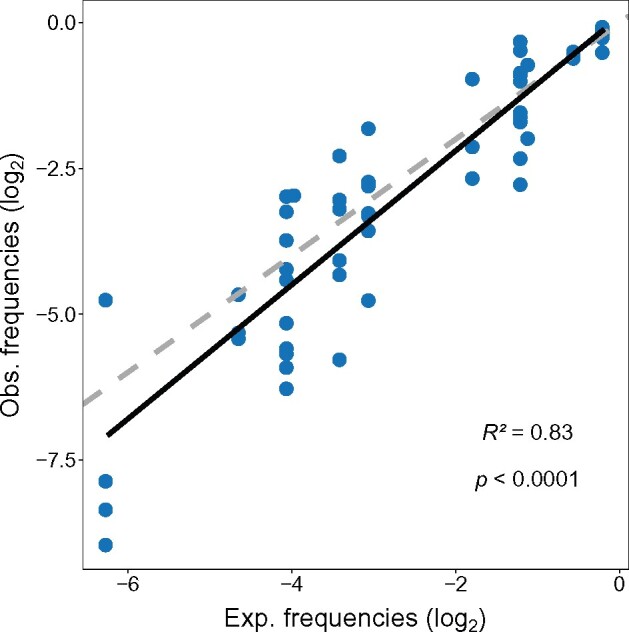
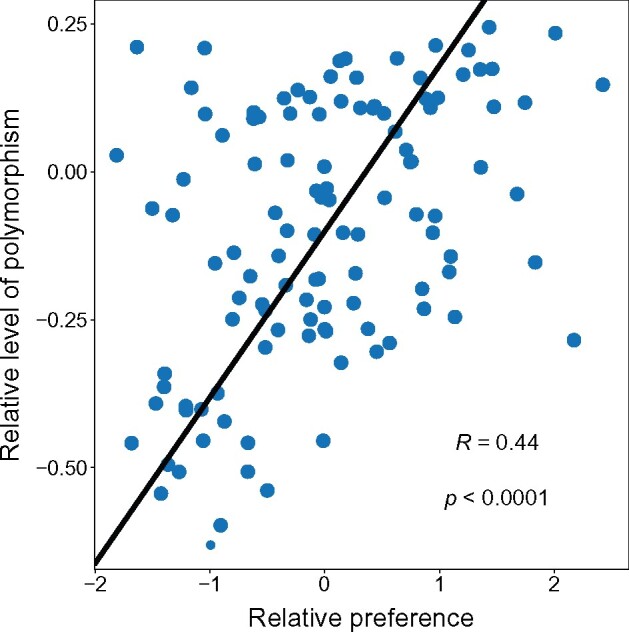
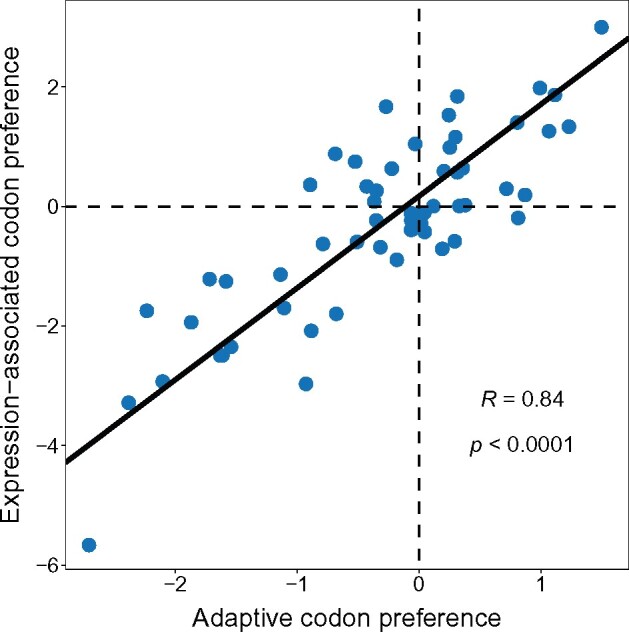
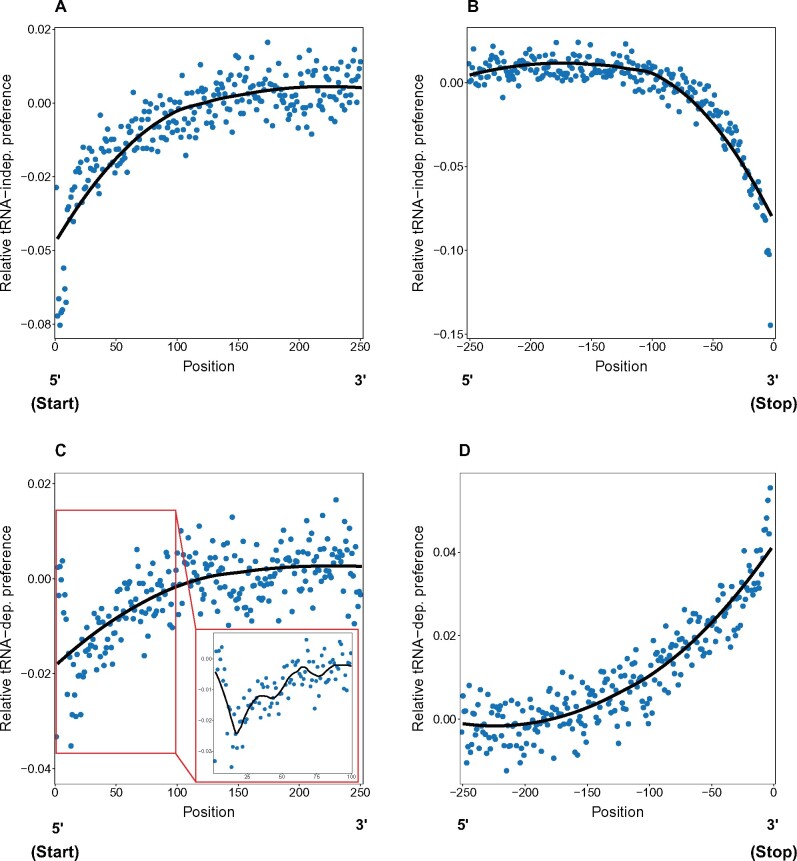
Alternative synonymous codons are often used at unequal frequencies. Classically, studies of such codon usage bias (CUB) attempted to separate the impact of neutral from selective forces by assuming that deviations from a predicted neutral equilibrium capture selection. However, GC-biased gene conversion (gBGC) can also cause deviation from a neutral null. Alternatively, selection has been inferred from CUB in highly expressed genes, but the accuracy of this approach has not been extensively tested, and gBGC can interfere with such extrapolations (e.g., if expression and gene conversion rates covary). It is therefore critical to examine deviations from a mutational null in a species with no gBGC. To achieve this goal, we implement such an analysis in the highly AT rich genome of Dictyostelium discoideum, where we find no evidence of gBGC. We infer neutral CUB under mutational equilibrium to quantify "adaptive codon preference," a nontautologous genome wide quantitative measure of the relative selection strength driving CUB. We observe signatures of purifying selection consistent with selection favoring adaptive codon preference. Preferred codons are not GC rich, underscoring the independence from gBGC. Expression-associated "preference" largely matches adaptive codon preference but does not wholly capture the influence of selection shaping patterns across all genes, suggesting selective constraints associated specifically with high expression. We observe patterns consistent with effects on mRNA translation and stability shaping adaptive codon preference. Thus, our approach to quantifying adaptive codon preference provides a framework for inferring the sources of selection that shape CUB across different contexts within the genome.
Keywords: biased gene conversion; codon usage bias; translation; weak selection.
© The Author(s) 2021. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
