

Protein Footprinting via Covalent Protein Painting Reveals Structural Changes of the Proteome in Alzheimer's Disease
- PMID: 33872013
- PMCID: PMC8477671
- DOI: 10.1021/acs.jproteome.0c00912
Protein Footprinting via Covalent Protein Painting Reveals Structural Changes of the Proteome in Alzheimer's Disease
Abstract
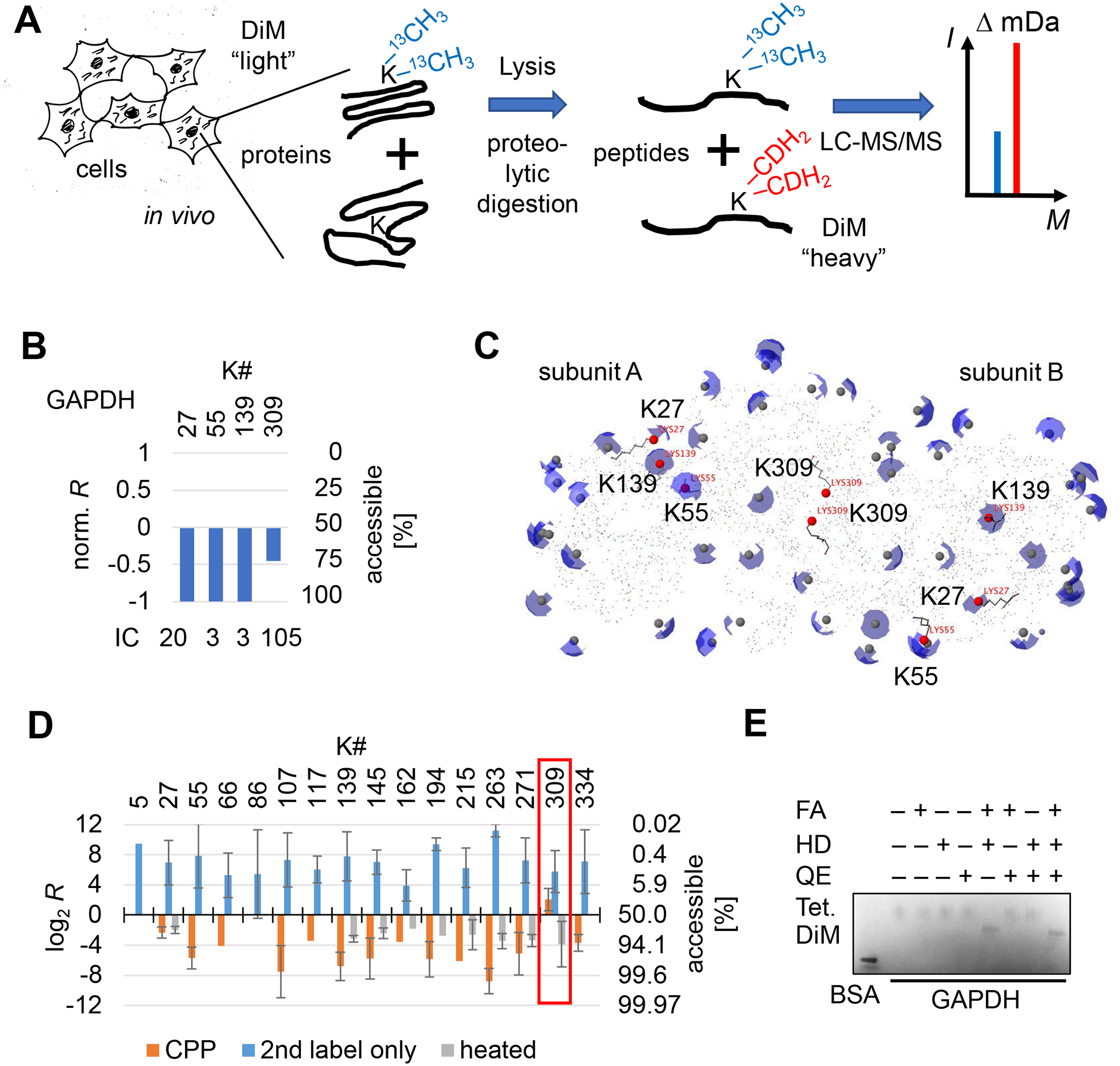
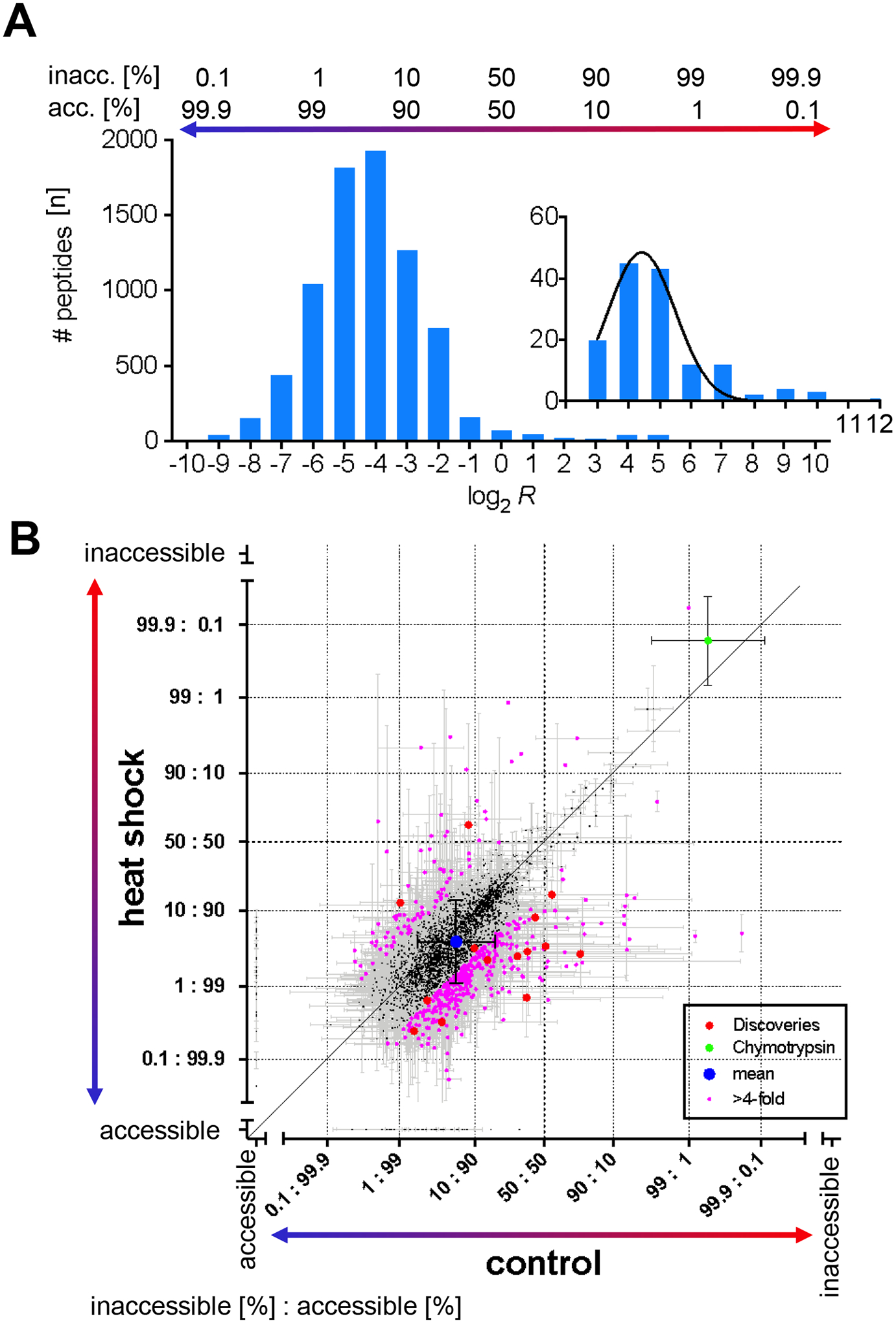
Misfolding and aggregation of amyloid-β peptide and hyperphosphorylated tau are molecular markers of Alzheimer's disease (AD), and although the 3D structures of these aberrantly folded proteins have been visualized in exquisite detail, no method has been able to survey protein folding across the proteome in AD. Here, we present covalent protein painting (CPP), a mass spectrometry-based protein footprinting approach to quantify the accessibility of lysine ε-amines for covalent modification at the surface of natively folded proteins. We used CPP to survey the reactivity of 2645 lysine residues and therewith the structural proteome of HEK293T cells and found that reactivity increased upon mild heat shock. CPP revealed that the accessibility of lysine residues for covalent modification in tubulin-β (TUBB), in succinate dehydrogenase (SHDB), and in amyloid-β peptide (Aβ) is altered in human postmortem brain samples of patients with neurodegenerative diseases. The structural alterations of TUBB and SHDB in patients with AD, dementia with Lewy bodies (DLB), or both point to broader perturbations of the 3D proteome beyond Aβ and hyperphosphorylated tau.
Keywords: MudPIT; bottom-up proteomics; chemical footprinting; conformational diagnostics; diffuse Lewy body disease; isobaric isotopologue; molecular diagnostics; neurodegenerative diseases; protein surface mapping; quantitative mass spectrometry; structural proteomics.
Conflict of interest statement
Competing financial interests
The authors do not declare competing financial interests.
Figures
Similar articles
-
Microglia phenotypes are associated with subregional patterns of concomitant tau, amyloid-β and α-synuclein pathologies in the hippocampus of patients with Alzheimer's disease and dementia with Lewy bodies.Acta Neuropathol Commun. 2022 Mar 16;10(1):36. doi: 10.1186/s40478-022-01342-7. Acta Neuropathol Commun. 2022. PMID: 35296366 Free PMC article.
-
CSF amyloid-beta-peptides in Alzheimer's disease, dementia with Lewy bodies and Parkinson's disease dementia.Brain. 2006 May;129(Pt 5):1177-87. doi: 10.1093/brain/awl063. Epub 2006 Apr 6. Brain. 2006. PMID: 16600985 Free PMC article.
-
Exploring the Role of Aggregated Proteomes in the Pathogenesis of Alzheimer's Disease.Curr Protein Pept Sci. 2020;21(12):1164-1173. doi: 10.2174/1389203721666200921152246. Curr Protein Pept Sci. 2020. PMID: 32957903 Review.
-
Global Profiling of Lysine Accessibility to Evaluate Protein Structure Changes in Alzheimer's Disease.J Am Soc Mass Spectrom. 2021 Apr 7;32(4):936-945. doi: 10.1021/jasms.0c00450. Epub 2021 Mar 8. J Am Soc Mass Spectrom. 2021. PMID: 33683887 Free PMC article.
-
Development of molecular tools for diagnosis of Alzheimer's disease that are based on detection of amyloidogenic proteins.Prion. 2021 Dec;15(1):56-69. doi: 10.1080/19336896.2021.1917289. Prion. 2021. PMID: 33910450 Free PMC article. Review.
Cited by
-
Quantitative structural proteomics in living cells by covalent protein painting.Methods Enzymol. 2023;679:33-63. doi: 10.1016/bs.mie.2022.08.046. Epub 2023 Jan 4. Methods Enzymol. 2023. PMID: 36682868 Free PMC article.
-
Cellular Exposure to Chloroacetanilide Herbicides Induces Distinct Protein Destabilization Profiles.ACS Chem Biol. 2023 Jul 21;18(7):1661-1676. doi: 10.1021/acschembio.3c00338. Epub 2023 Jul 10. ACS Chem Biol. 2023. PMID: 37427419 Free PMC article.
-
Protein painting for structural and binding site analysis via intracellular lysine reactivity profiling with o-phthalaldehyde.Chem Sci. 2024 Mar 20;15(16):6064-6075. doi: 10.1039/d4sc00032c. eCollection 2024 Apr 24. Chem Sci. 2024. PMID: 38665522 Free PMC article.
-
Hsp40 Affinity to Identify Proteins Destabilized by Cellular Toxicant Exposure.Anal Chem. 2021 Dec 21;93(50):16940-16946. doi: 10.1021/acs.analchem.1c04230. Epub 2021 Dec 7. Anal Chem. 2021. PMID: 34874156 Free PMC article.
-
Comprehensive Identification of Regulatory Protein Networks.J Proteome Res. 2021 Nov 5;20(11):4913-4914. doi: 10.1021/acs.jproteome.1c00813. J Proteome Res. 2021. PMID: 34736325 Free PMC article. No abstract available.
References
-
- Scheltens P et al.Alzheimer’s disease. Lancet 388, 505–517, (2016). - PubMed
-
- Balch WE, Morimoto RI, Dillin A & Kelly JW Adapting proteostasis for disease intervention. Science 319, 916–919, (2008). - PubMed
-
- Kallen RG & Jencks WP Equilibria for the reaction of amines with formaldehyde and protons in aqueous solution. A re-examination of the formol titration. The Journal of biological chemistry 241, 5864–5878, (1966). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
