

Case Reports
doi: 10.7326/L21-0108.
Epub 2021 Apr 20.
SARS-CoV-2 Reinfection in a Liver Transplant Recipient
Affiliations
- PMID: 33872044
- PMCID: PMC8059415
- DOI: 10.7326/L21-0108
Item in Clipboard
Case Reports
SARS-CoV-2 Reinfection in a Liver Transplant Recipient
Ann Intern Med.
2021 Aug.
No abstract available
Conflict of interest statement
Figures
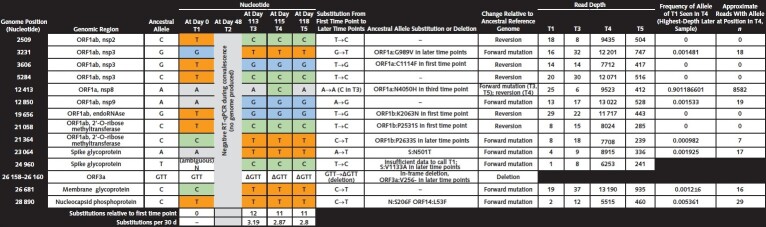
Five nasopharyngeal specimens were collected spanning 118 d. For viral sequencing, 2 replicate sequencing libraries were prepared from source material for each sample as previously described (1). SARS-CoV-2 genomes were assembled using viral-ngs, v2.1.10.0, assembly pipelines (2). Consensus SARS-CoV-2 genomes were assembled for all positive time points, whereas no genomic data were produced from the negative RT-qPCR test result (T2). The genome from the first time point was 96.6% complete (mean depth: 18 reads), and remaining genomes were 99% complete (mean depth: tens to thousands of reads). Each assembled genome was characterized by comparison to the ancestral reference genome, NC_045512.2 (isolated from one of the first known COVID-19 cases in Wuhan, China). The 3 later time points are nearly identical and share a common set of single-nucleotide variants (SNVs), with T3
having a single additional SNV. Compared with T1, these 3 genomes had more substitutions (11-12 SNVs) than expected from the mean substitution rate of SARS-CoV-2, which is approximately 1 substitution every 2 wk (3). Of note, 5 of the substitutions seen in the first time point were replaced by the ancestral allele in later time points; these apparent reversions strongly suggest that the later genomes reflect an independent infection with a virus from a distinct lineage rather than evolution of the virus of the first time point, especially given the ubiquity of SARS-CoV-2 in the surrounding community. Three amino acid changes present at the first time point were absent from the later time points, and the later time points all bear 3 new amino acid substitutions not seen in the first time point, as well as a deletion. Time points T3–T5 had a notable amino acid substitution in the receptor-binding domain of the
spike glycoprotein at position 501 (S:N501T), an amino acid substitution believed to increase affinity for the angiotensin-converting enzyme 2 receptor (4). In the most deeply sequenced later time point, T4, none of the distinguishing variants of the first time point were present in high abundance, and nearly half were absent entirely. For none of the apparent reversions to the ancestral allele did a minor population exist in the most densely sequenced later time point, T4. RT-qPCR = reverse transcriptase quantitative polymerase chain reaction.
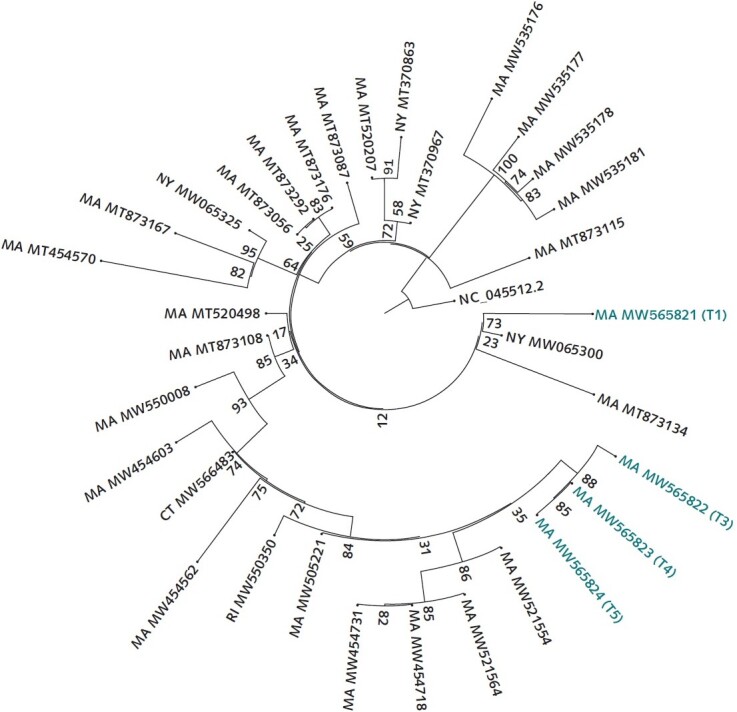
Similarity by genetic distance and placement on the phylogenetic tree show that the second infection is more closely related to and descended from infections circulating in the community than from the viral genome sequenced from the first infection. The sequences were aligned to the reference NC_045512.2 using MAFFT. A maximum likelihood tree was created via IQ-Tree with the general time-reversible model with empirical base frequencies and 3 FreeRate categories, as selected by minimum Akaike information criterion. CT = Connecticut; MA = Massachusetts; ME = Maine; NH = New Hampshire; NY = New York; RI = Rhode Island; VT = Vermont.
Comment in
-
COVID-19 Reinfection by the Gamma Variant in Kidney Transplant Recipients.Transplantation. 2021 Dec 1;105(12):e276-e277. doi: 10.1097/TP.0000000000003924. Transplantation. 2021. PMID: 34392271 Free PMC article. No abstract available.
References
-
- Centers for Disease Control and Prevention. Common investigation protocol for investigating suspected SARS-CoV-2 reinfection. 27 October 2020. Accessed at www.cdc.gov/coronavirus/2019-ncov/php/reinfection.html on 23 November 2020.
-
- viral-pipelines. GitHub. Accessed at https://github.com/broadinstitute/viral-pipelines on 22 March 2021.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous