

Role of microglial and endothelial CD36 in post-ischemic inflammasome activation and interleukin-1β-induced endothelial activation
- PMID: 33872708
- PMCID: PMC8187325
- DOI: 10.1016/j.bbi.2021.04.010
Role of microglial and endothelial CD36 in post-ischemic inflammasome activation and interleukin-1β-induced endothelial activation
Abstract
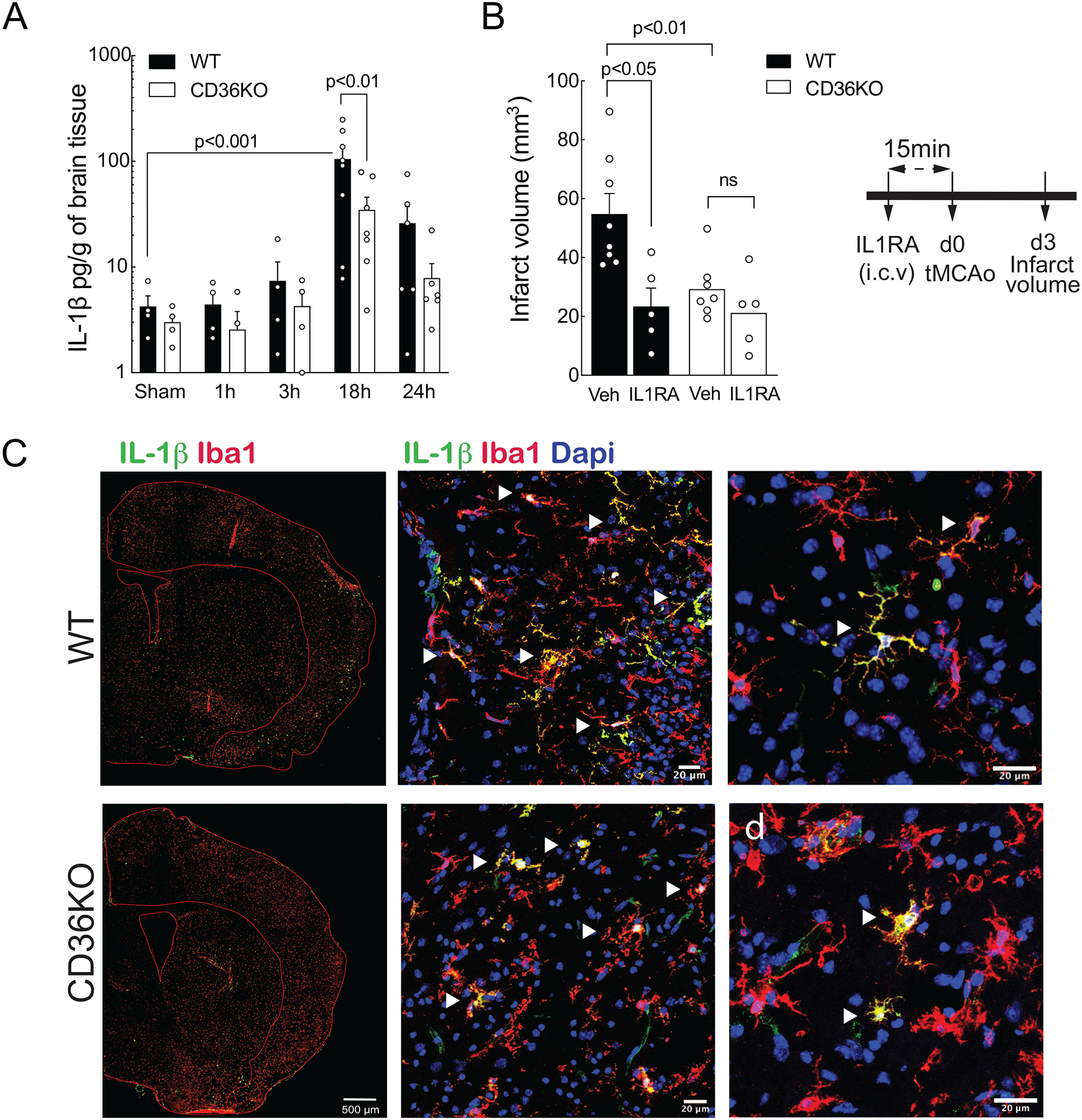
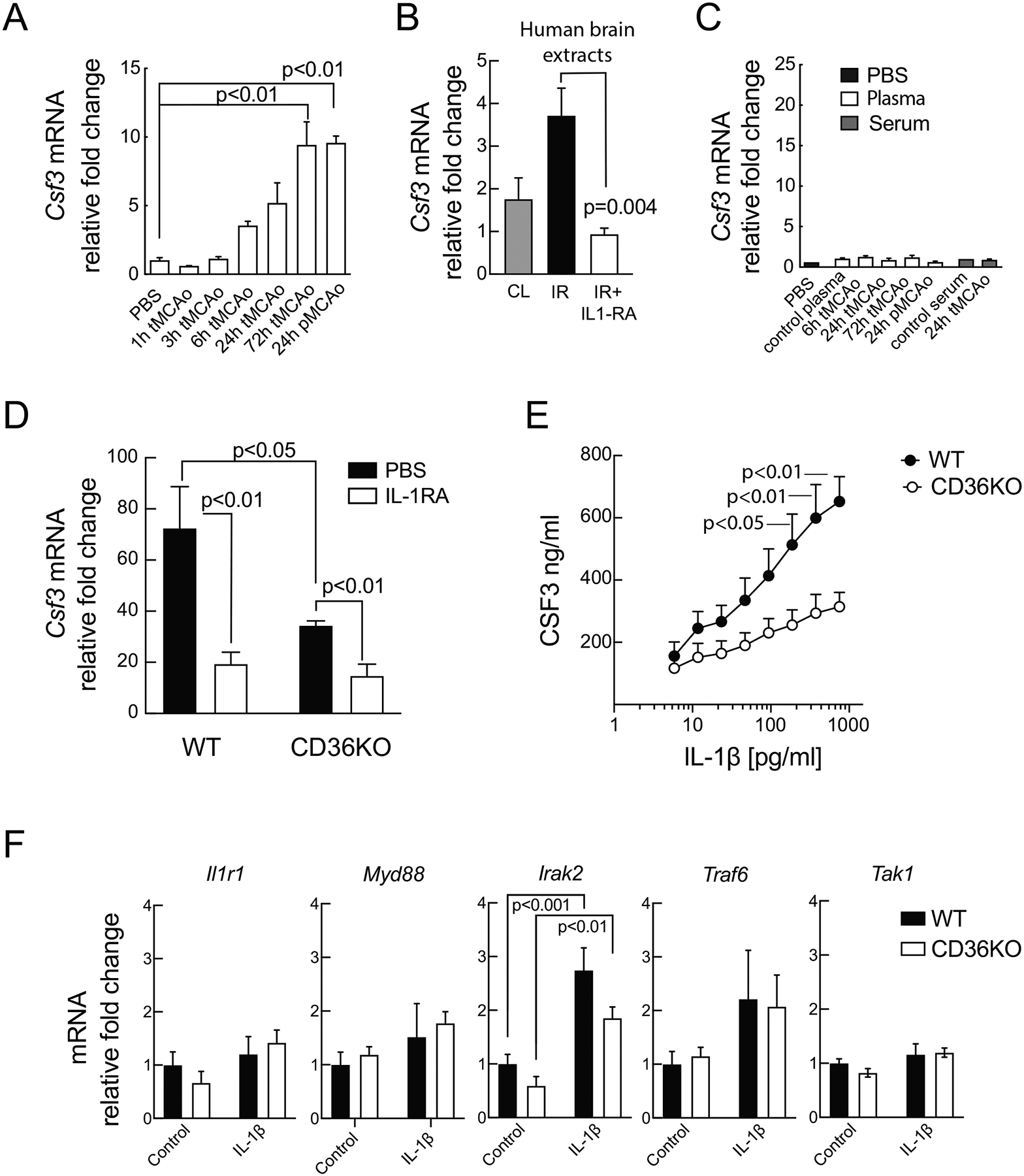
Cerebral ischemia is associated with an acute inflammatory response that contributes to the resulting injury. The innate immunity receptor CD36, expressed in microglia and endothelium, and the pro-inflammatory cytokine interleukin-1β (IL-1β) are involved in the mechanisms of ischemic injury. Since CD36 has been implicated in activation of the inflammasome, the main source of IL-1β, we investigated whether CD36 mediates brain injury through the inflammasome and IL-1β. We found that active caspase-1, a key inflammasome component, is decreased in microglia of CD36-deficient mice subjected to transient middle cerebral artery occlusion, an effect associated with a reduction in brain IL-1β. Conditional deletion of CD36 either in microglia or endothelium reduced ischemic injury in mice, attesting to the pathogenic involvement of CD36 in both cell types. Application of an ischemic brain extract to primary brain endothelial cell cultures from wild type (WT) mice induced IL-1β-dependent endothelial activation, reflected by increases in the cytokine colony stimulating factor-3, a response markedly attenuated in CD36-deficient endothelia. Similarly, the increase in colony stimulating factor-3 induced by recombinant IL-1β was attenuated in CD36-deficient compared to WT endothelia. We conclude that microglial CD36 is a key determinant of post-ischemic IL-1β production by regulating caspase-1 activity, whereas endothelial CD36 is required for the full expression of the endothelial activation induced by IL-1β. The data identify microglial and endothelial CD36 as critical upstream components of the acute inflammatory response to cerebral ischemia and viable putative therapeutic targets.
Keywords: Caspase-1; Csf3; IL-1β; Neuroinflammation; Pattern recognition receptors; Stroke.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflicts of interest to declare
CI serves on the Scientific Advisory Board of Broadview Ventures. The other authors have no conflict of interest to declare.
Figures



References
-
- Abulafia DP, de Rivero Vaccari JP, Lozano JD, Lotocki G, Keane RW, Dietrich WD, 2009. Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J Cereb Blood Flow Metab 29, 534–544. - PubMed
-
- Allan SM, Tyrrell PJ, Rothwell NJ, 2005. Interleukin-1 and neuronal injury. Nat Rev Immunol 5, 629–640. - PubMed
-
- Amantea D, Nappi G, Bernardi G, Bagetta G, Corasaniti MT, 2009. Postischemic brain damage: pathophysiology and role of inflammatory mediators. FEBS J 276, 13–26. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
