

Rare deleterious mutations of HNRNP genes result in shared neurodevelopmental disorders
- PMID: 33874999
- PMCID: PMC8056596
- DOI: 10.1186/s13073-021-00870-6
Rare deleterious mutations of HNRNP genes result in shared neurodevelopmental disorders
Abstract
Background: With the increasing number of genomic sequencing studies, hundreds of genes have been implicated in neurodevelopmental disorders (NDDs). The rate of gene discovery far outpaces our understanding of genotype-phenotype correlations, with clinical characterization remaining a bottleneck for understanding NDDs. Most disease-associated Mendelian genes are members of gene families, and we hypothesize that those with related molecular function share clinical presentations.
Methods: We tested our hypothesis by considering gene families that have multiple members with an enrichment of de novo variants among NDDs, as determined by previous meta-analyses. One of these gene families is the heterogeneous nuclear ribonucleoproteins (hnRNPs), which has 33 members, five of which have been recently identified as NDD genes (HNRNPK, HNRNPU, HNRNPH1, HNRNPH2, and HNRNPR) and two of which have significant enrichment in our previous meta-analysis of probands with NDDs (HNRNPU and SYNCRIP). Utilizing protein homology, mutation analyses, gene expression analyses, and phenotypic characterization, we provide evidence for variation in 12 HNRNP genes as candidates for NDDs. Seven are potentially novel while the remaining genes in the family likely do not significantly contribute to NDD risk.
Results: We report 119 new NDD cases (64 de novo variants) through sequencing and international collaborations and combined with published clinical case reports. We consider 235 cases with gene-disruptive single-nucleotide variants or indels and 15 cases with small copy number variants. Three hnRNP-encoding genes reach nominal or exome-wide significance for de novo variant enrichment, while nine are candidates for pathogenic mutations. Comparison of HNRNP gene expression shows a pattern consistent with a role in cerebral cortical development with enriched expression among radial glial progenitors. Clinical assessment of probands (n = 188-221) expands the phenotypes associated with HNRNP rare variants, and phenotypes associated with variation in the HNRNP genes distinguishes them as a subgroup of NDDs.
Conclusions: Overall, our novel approach of exploiting gene families in NDDs identifies new HNRNP-related disorders, expands the phenotypes of known HNRNP-related disorders, strongly implicates disruption of the hnRNPs as a whole in NDDs, and supports that NDD subtypes likely have shared molecular pathogenesis. To date, this is the first study to identify novel genetic disorders based on the presence of disorders in related genes. We also perform the first phenotypic analyses focusing on related genes. Finally, we show that radial glial expression of these genes is likely critical during neurodevelopment. This is important for diagnostics, as well as developing strategies to best study these genes for the development of therapeutics.
Keywords: Cortex development; Gene families; Neurodevelopmental disorders; hnRNPs.
Conflict of interest statement
The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing conducted at Baylor Genetics Laboratory. E.T. is an employee of GeneDx. The remaining authors declare no competing interests.
Figures
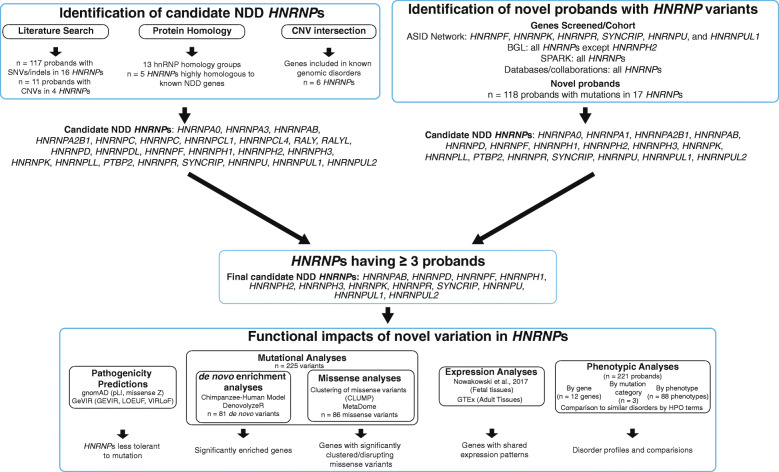




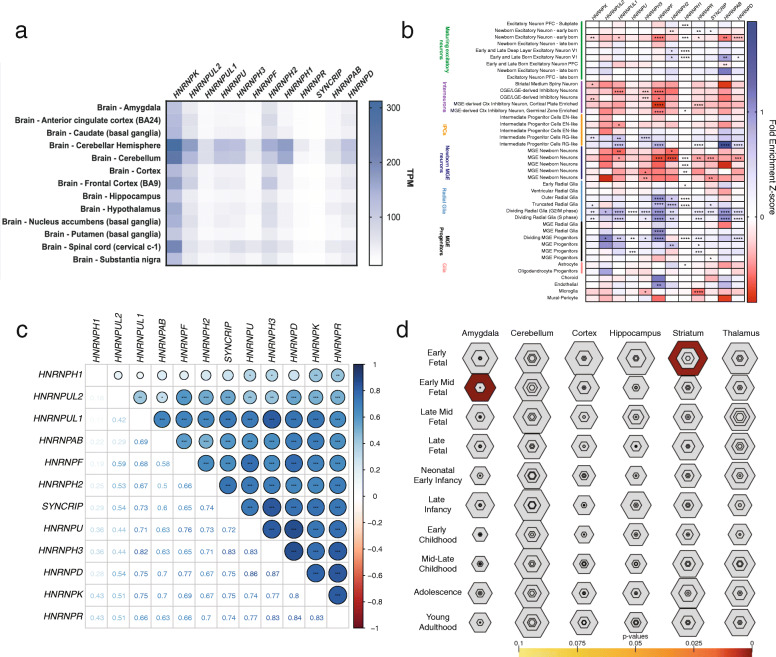


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
