

Biallelic variants in the SORD gene are one of the most common causes of hereditary neuropathy among Czech patients
- PMID: 33875678
- PMCID: PMC8055917
- DOI: 10.1038/s41598-021-86857-0
Biallelic variants in the SORD gene are one of the most common causes of hereditary neuropathy among Czech patients
Abstract
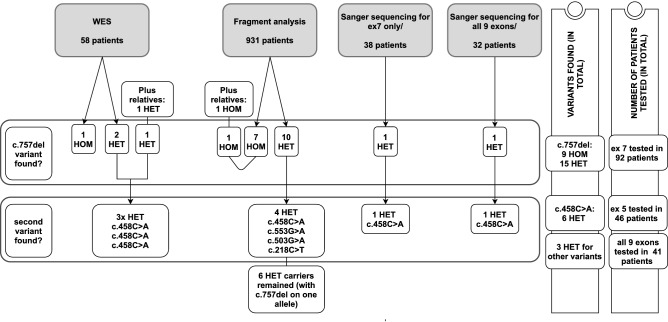
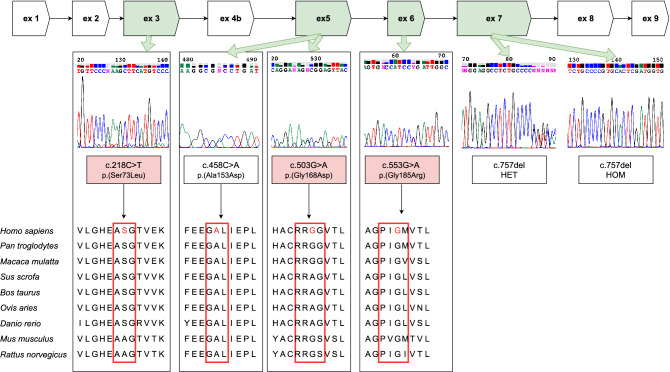
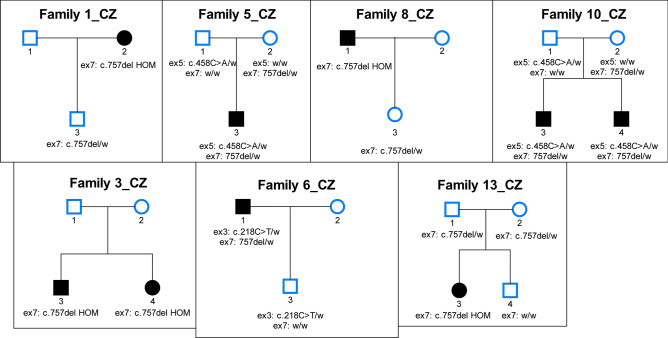
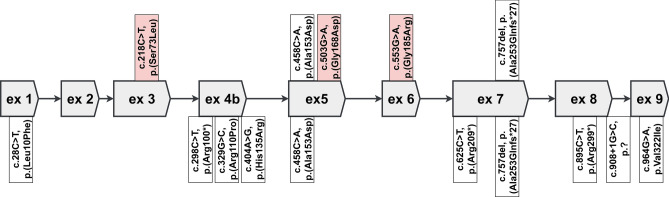
Recently, biallelic variants in the SORD gene were identified as causal for axonal hereditary neuropathy (HN). We ascertained the spectrum and frequency of SORD variants among a large cohort of Czech patients with unknown cause of HN. Exome sequencing data were analysed for SORD (58 patients). The prevalent c.757del variant was tested with fragment analysis (931 patients). Sanger sequencing in additional 70 patients was done. PCR primers were designed to amplify the SORD gene with the exclusion of the pseudogene SORD2P. Sequence differences between gene and pseudogene were identified and frequencies of SNPs were calculated. Eighteen patients from 16 unrelated families with biallelic variants in the SORD gene were found and the c.757del was present in all patients on at least one allele. Three novel, probably pathogenic, variants were detected, always in a heterozygous state in combination with the c.757del on the second allele. Patients presented with a slowly progressive axonal HN. Almost all patients had moderate pes cavus deformity. SORD neuropathy is frequent in Czech patients and the third most common cause of autosomal recessive HN. The c.757del is highly prevalent. Specific amplification of the SORD gene with the exclusion of the pseudogene is essential for a precise molecular diagnostics.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Mendoza-Ferreira N, Karakaya M, Cengiz N, Beijer D, Brigatti KW, Gonzaga-Jauregui C, Fuhrmann N, Holker I, Thelen MP, Zetzsche S, Rombo R, Puffenberger EG, De Jonghe P, Deconinck T, Zuchner S, Strauss KA, Carson V, Schrank B, Wunderlich G, Baets J, Wirth B. De Novo and inherited variants in GBF1 are associated with axonal neuropathy caused by golgi fragmentation. Am. J. Hum. Genet. 2020;107:763–777. doi: 10.1016/j.ajhg.2020.08.018. - DOI - PMC - PubMed
-
- Cortese A, Zhu Y, Rebelo AP, Negri S, Courel S, Abreu L, Bacon CJ, Bai Y, Bis-Brewer DM, Bugiardini E, Buglo E, Danzi MC, Feely SME, Athanasiou-Fragkouli A, Haridy NA, Inherited Neuropathy C, Isasi R, Khan A, Laura M, Magri S, Pipis M, Pisciotta C, Powell E, Rossor AM, Saveri P, Sowden JE, Tozza S, Vandrovcova J, Dallman J, Grignani E, Marchioni E, Scherer SS, Tang B, Lin Z, Al-Ajmi A, Schule R, Synofzik M, Maisonobe T, Stojkovic T, Auer-Grumbach M, Abdelhamed MA, Hamed SA, Zhang R, Manganelli F, Santoro L, Taroni F, Pareyson D, Houlden H, Herrmann DN, Reilly MM, Shy ME, Zhai RG, Zuchner S. Biallelic mutations in SORD cause a common and potentially treatable hereditary neuropathy with implications for diabetes. Nat. Genet. 2020;52:473–481. doi: 10.1038/s41588-020-0615-4. - DOI - PMC - PubMed
-
- Azzedine H, Zavadakova P, Plante-Bordeneuve V, Vaz Pato M, Pinto N, Bartesaghi L, Zenker J, Poirot O, Bernard-Marissal N, Arnaud Gouttenoire E, Cartoni R, Title A, Venturini G, Medard JJ, Makowski E, Schols L, Claeys KG, Stendel C, Roos A, Weis J, Dubourg O, Leal Loureiro J, Stevanin G, Said G, Amato A, Baraban J, LeGuern E, Senderek J, Rivolta C, Chrast R. PLEKHG5 deficiency leads to an intermediate form of autosomal-recessive Charcot-Marie-Tooth disease. Hum. Mol. Genet. 2013;22:4224–4232. doi: 10.1093/hmg/ddt274. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
