

Combining exome/genome sequencing with data repository analysis reveals novel gene-disease associations for a wide range of genetic disorders
- PMID: 33875846
- PMCID: PMC8354858
- DOI: 10.1038/s41436-021-01159-0
Combining exome/genome sequencing with data repository analysis reveals novel gene-disease associations for a wide range of genetic disorders
Abstract
Purpose: Within this study, we aimed to discover novel gene-disease associations in patients with no genetic diagnosis after exome/genome sequencing (ES/GS).
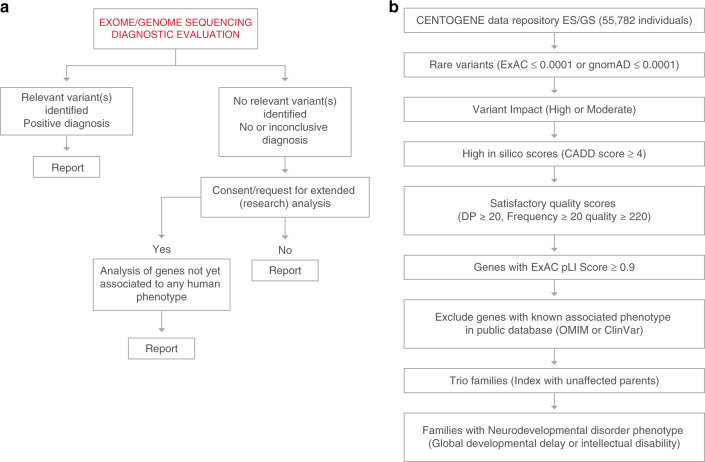
Methods: We followed two approaches: (1) a patient-centered approach, which after routine diagnostic analysis systematically interrogates variants in genes not yet associated to human diseases; and (2) a gene variant centered approach. For the latter, we focused on de novo variants in patients that presented with neurodevelopmental delay (NDD) and/or intellectual disability (ID), which are the most common reasons for genetic testing referrals. Gene-disease association was assessed using our data repository that combines ES/GS data and Human Phenotype Ontology terms from over 33,000 patients.
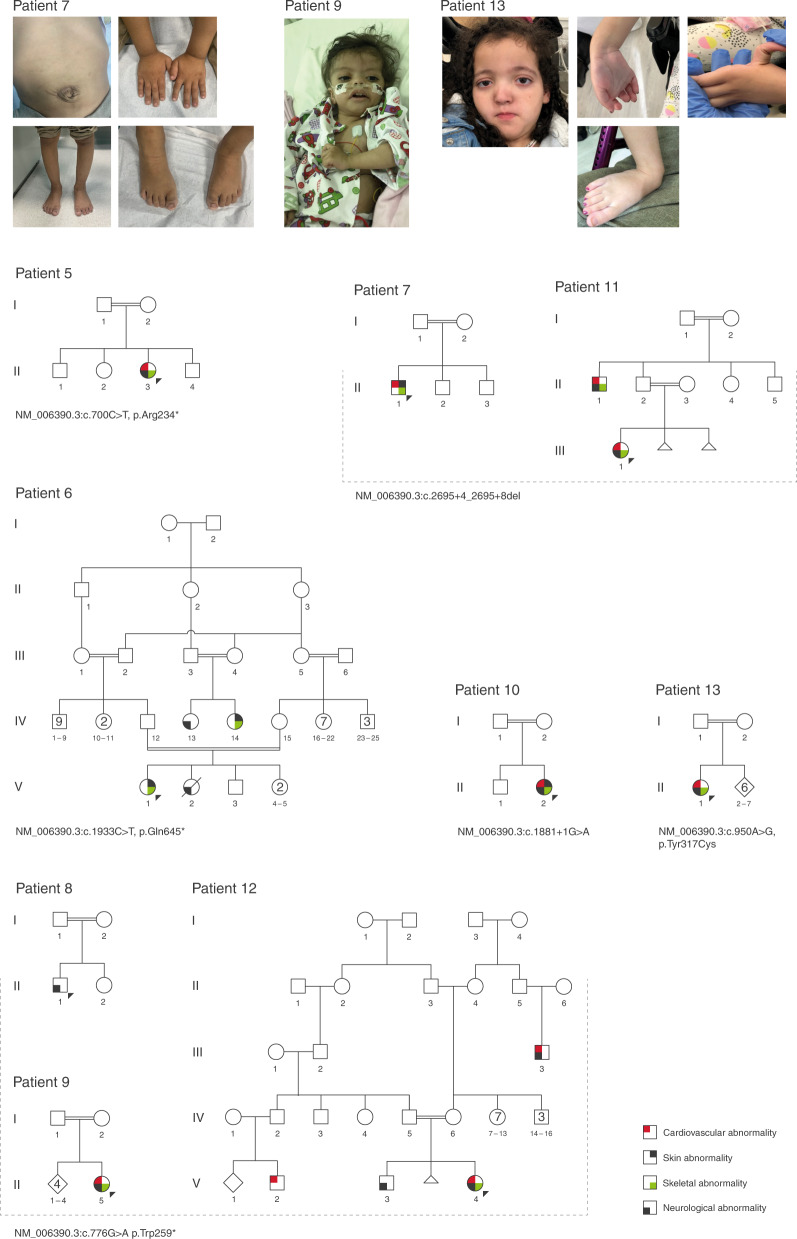
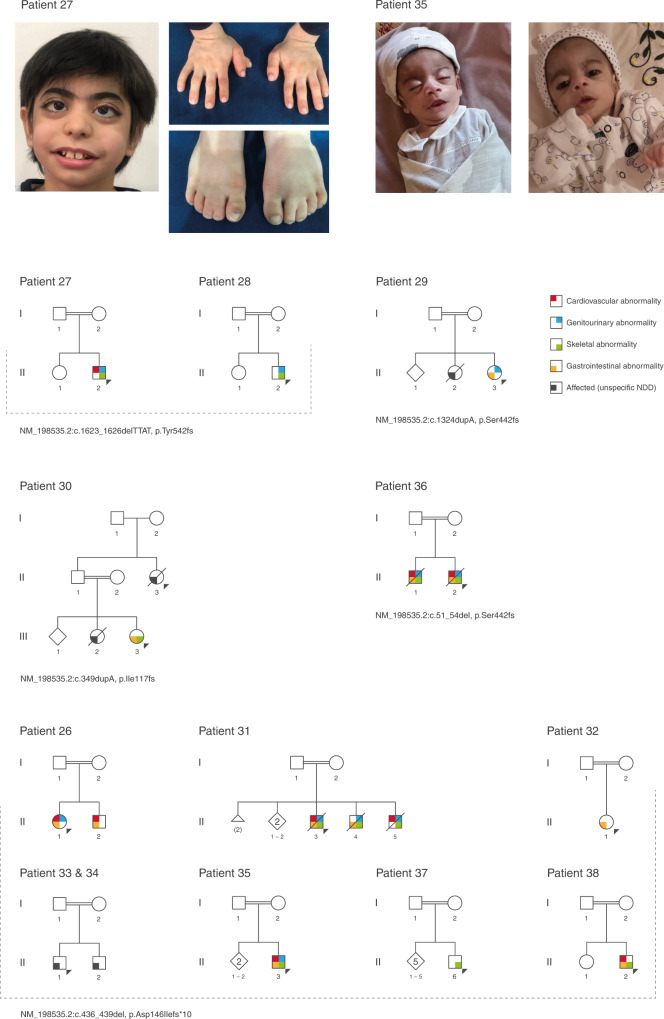
Results: We propose six novel gene-disease associations based on 38 patients with variants in the BLOC1S1, IPO8, MMP15, PLK1, RAP1GDS1, and ZNF699 genes. Furthermore, our results support causality of 31 additional candidate genes that had little published evidence and no registered OMIM phenotype (56 patients). The phenotypes included syndromic/nonsyndromic NDD/ID, oral-facial-digital syndrome, cardiomyopathies, malformation syndrome, short stature, skeletal dysplasia, and ciliary dyskinesia.
Conclusion: Our results demonstrate the value of data repositories which combine clinical and genetic data for discovering and confirming gene-disease associations. Genetic laboratories should be encouraged to pursue such analyses for the benefit of undiagnosed patients and their families.
© 2021. The Author(s).
Conflict of interest statement
A.M.B.-A., K.K.K., S.K., N.O.-H., N.O.-H., K.T., C.B., M.E.R., A.U., R.H., A.L., R.A.-A., V.K., N.A., and P.B. are employees of CENTOGENE GmbH. A.R. is the former CEO of CENTOGENE GmbH. The other authors declare no competing interests.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
