

Lipid-mediated motor-adaptor sequestration impairs axonal lysosome delivery leading to autophagic stress and dystrophy in Niemann-Pick type C
- PMID: 33878344
- PMCID: PMC8137671
- DOI: 10.1016/j.devcel.2021.03.032
Lipid-mediated motor-adaptor sequestration impairs axonal lysosome delivery leading to autophagic stress and dystrophy in Niemann-Pick type C
Abstract
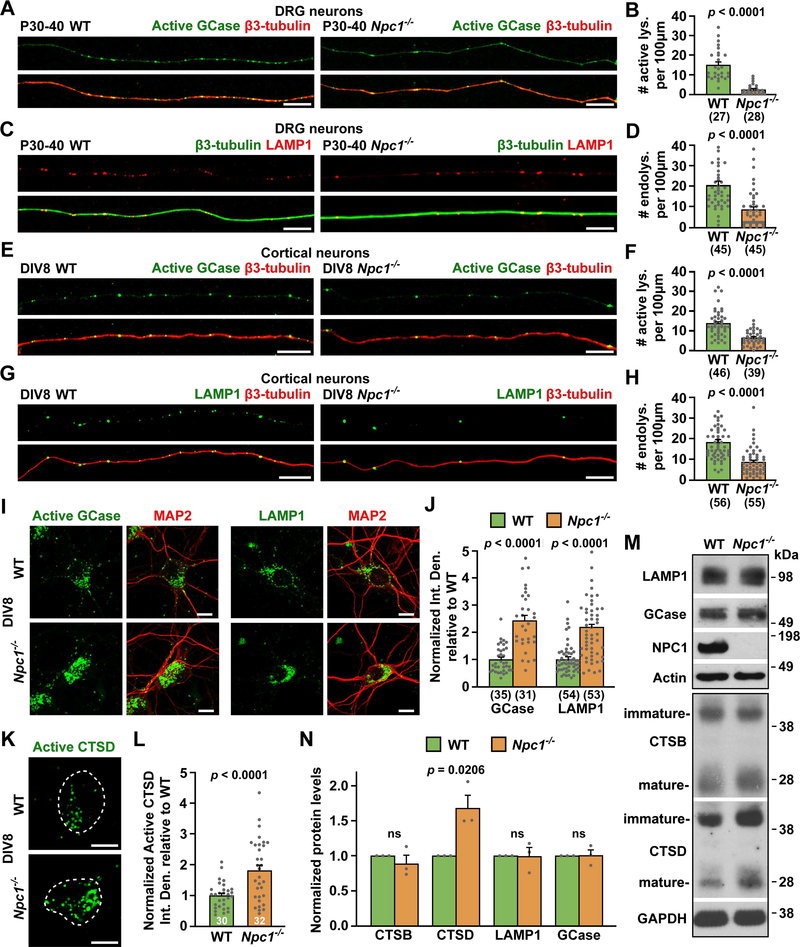
Niemann-Pick disease type C (NPC) is a neurodegenerative lysosomal storage disorder characterized by lipid accumulation in endolysosomes. An early pathologic hallmark is axonal dystrophy occurring at presymptomatic stages in NPC mice. However, the mechanisms underlying this pathologic change remain obscure. Here, we demonstrate that endocytic-autophagic organelles accumulate in NPC dystrophic axons. Using super-resolution and live-neuron imaging, we reveal that elevated cholesterol on NPC lysosome membranes sequesters kinesin-1 and Arl8 independent of SKIP and Arl8-GTPase activity, resulting in impaired lysosome transport into axons, contributing to axonal autophagosome accumulation. Pharmacologic reduction of lysosomal membrane cholesterol with 2-hydroxypropyl-β-cyclodextrin (HPCD) or elevated Arl8b expression rescues lysosome transport, thereby reducing axonal autophagic stress and neuron death in NPC. These findings demonstrate a pathological mechanism by which altered membrane lipid composition impairs lysosome delivery into axons and provide biological insights into the translational application of HPCD in restoring axonal homeostasis at early stages of NPC disease.
Keywords: Niemann-Pick disease type C; autophagosome; axonal dystrophy; axonal transport; cholesterol; kinesin; lipid; lysosomal storage disorder; lysosome; neurodegeneration.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures






Comment in
-
Lysosome transport interrupted.Nat Rev Mol Cell Biol. 2021 Jun;22(6):373. doi: 10.1038/s41580-021-00376-4. Nat Rev Mol Cell Biol. 2021. PMID: 33911233 No abstract available.
-
Lysosomes to the rescue: Anterograde axonal lysosome transport and neuronal proteostasis.Dev Cell. 2021 May 17;56(10):1361-1362. doi: 10.1016/j.devcel.2021.04.024. Dev Cell. 2021. PMID: 34004149
-
Lipid-mediated impairment of axonal lysosome transport contributing to autophagic stress.Autophagy. 2021 Jul;17(7):1796-1798. doi: 10.1080/15548627.2021.1938916. Epub 2021 Jun 30. Autophagy. 2021. PMID: 34085599 Free PMC article.
References
-
- Beard H, Hassiotis S, Gai WP, Parkinson-Lawrence E, Hopwood JJ, and Hemsley KM (2017). Axonal dystrophy in the brain of mice with Sanfilippo syndrome. Exp Neurol 295, 243–255. - PubMed
-
- Bilgin M, Nylandsted J, Jaattela M, and Maeda K (2017). Quantitative Profiling of Lysosomal Lipidome by Shotgun Lipidomics. Methods Mol Biol 1594, 19–34. - PubMed
-
- Boland B, and Platt FM (2015). Bridging the age spectrum of neurodegenerative storage diseases. Best Pract Res Clin Endocrinol Metab 29, 127–143. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
