

Genotype and defects in microtubule-based motility correlate with clinical severity in KIF1A-associated neurological disorder
- PMID: 33880452
- PMCID: PMC8054982
- DOI: 10.1016/j.xhgg.2021.100026
Genotype and defects in microtubule-based motility correlate with clinical severity in KIF1A-associated neurological disorder
Abstract
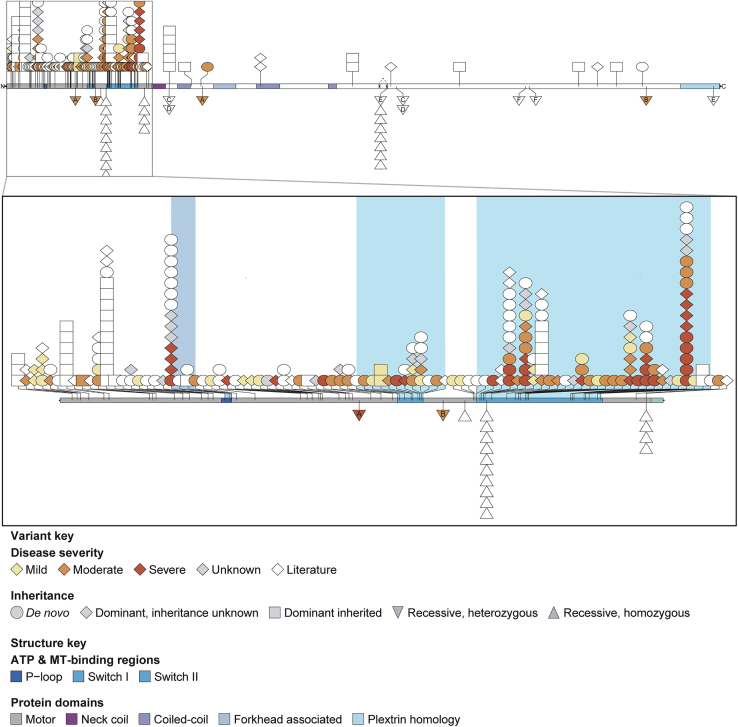
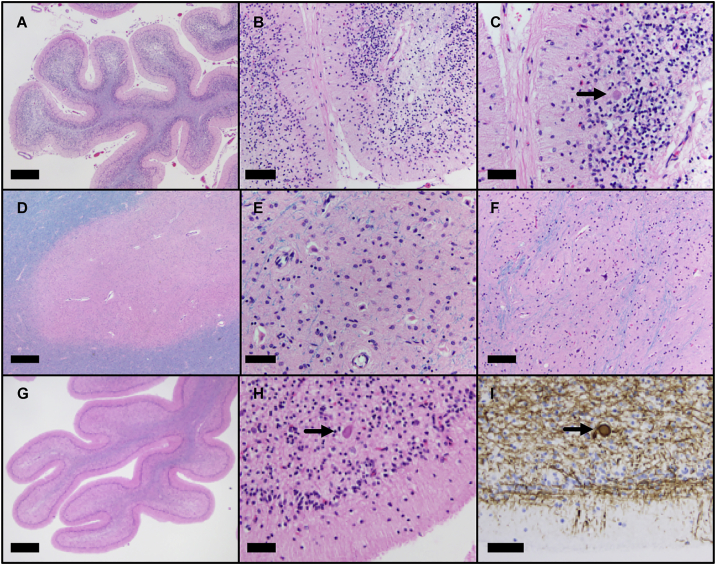
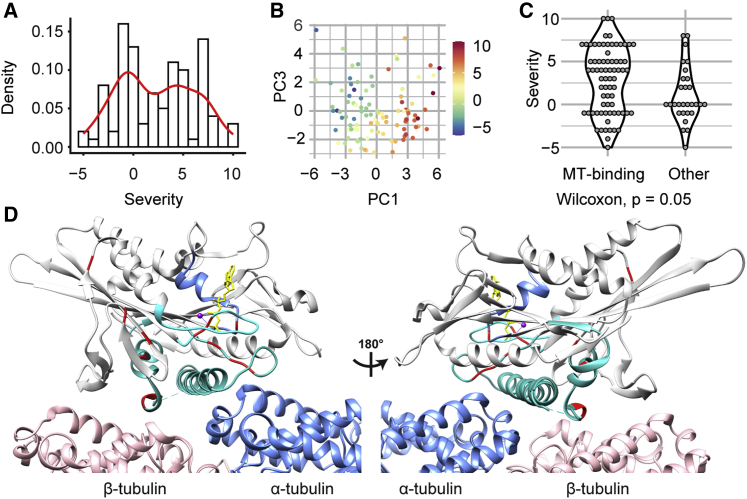
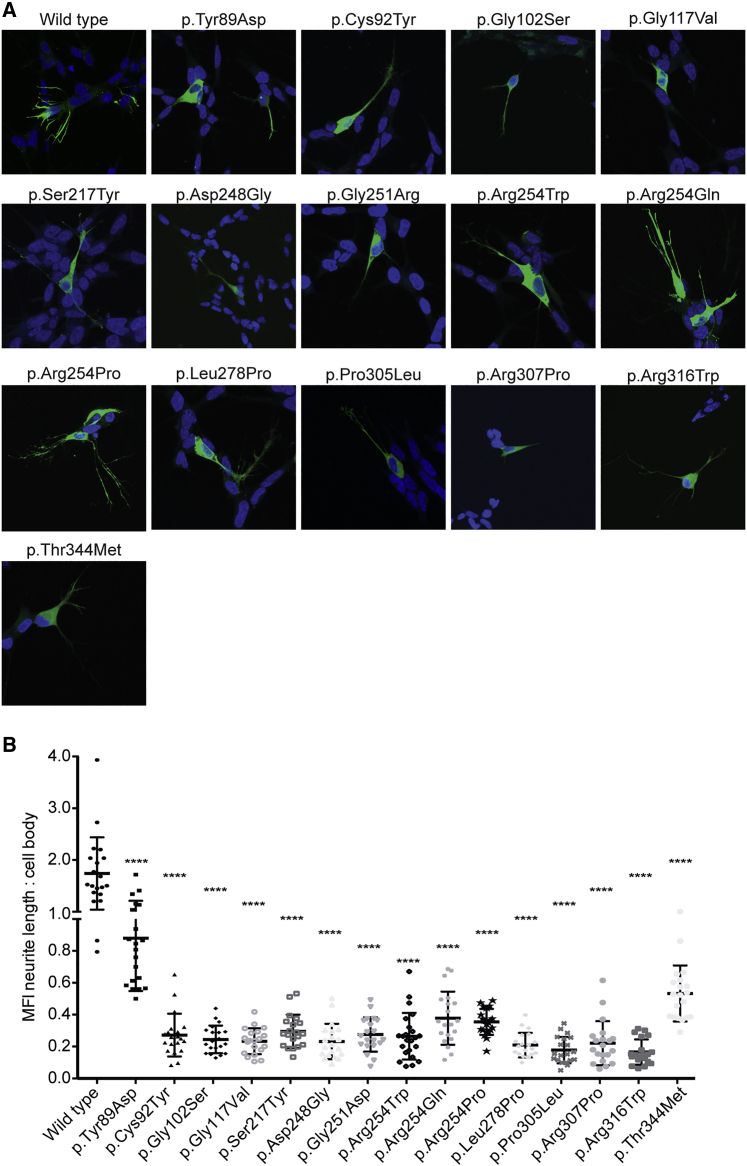
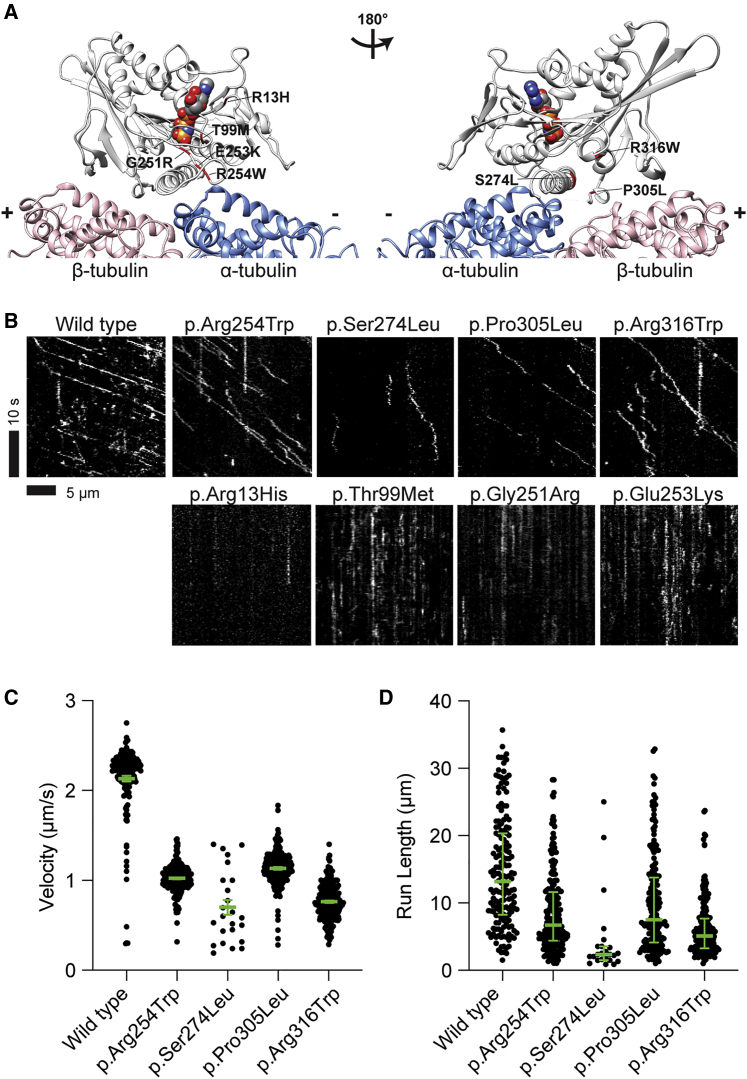
KIF1A-associated neurological disorder (KAND) encompasses a group of rare neurodegenerative conditions caused by variants in KIF1A,a gene that encodes an anterograde neuronal microtubule (MT) motor protein. Here we characterize the natural history of KAND in 117 individuals using a combination of caregiver or self-reported medical history, a standardized measure of adaptive behavior, clinical records, and neuropathology. We developed a heuristic severity score using a weighted sum of common symptoms to assess disease severity. Focusing on 100 individuals, we compared the average clinical severity score for each variant with in silico predictions of deleteriousness and location in the protein. We found increased severity is strongly associated with variants occurring in protein regions involved with ATP and MT binding: the P loop, switch I, and switch II. For a subset of variants, we generated recombinant proteins, which we used to assess transport in vivo by assessing neurite tip accumulation and to assess MT binding, motor velocity, and processivity using total internal reflection fluorescence microscopy. We find all modeled variants result in defects in protein transport, and we describe three classes of protein dysfunction: reduced MT binding, reduced velocity and processivity, and increased non-motile rigor MT binding. The rigor phenotype is consistently associated with the most severe clinical phenotype, while reduced MT binding is associated with milder clinical phenotypes. Our findings suggest the clinical phenotypic heterogeneity in KAND likely reflects and parallels diverse molecular phenotypes. We propose a different way to describe KAND subtypes to better capture the breadth of disease severity.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Alfadhel M., Yong S.L., Lillquist Y., Langlois S. Precocious puberty in two girls with PEHO syndrome: a clinical feature not previously described. J. Child Neurol. 2011;26:851–857. - PubMed
-
- Rivière J.-B., Ramalingam S., Lavastre V., Shekarabi M., Holbert S., Lafontaine J., Srour M., Merner N., Rochefort D., Hince P., et al. KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am. J. Hum. Genet. 2011;89:219–230. - PMC - PubMed
-
- Hamdan F.F., Gauthier J., Araki Y., Lin D.T., Yoshizawa Y., Higashi K., Park A.R., Spiegelman D., Dobrzeniecka S., Piton A., et al. S2D Group Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am. J. Hum. Genet. 2011;88:306–316. - PMC - PubMed
-
- Klebe S., Lossos A., Azzedine H., Mundwiller E., Sheffer R., Gaussen M., Marelli C., Nawara M., Carpentier W., Meyer V., et al. KIF1A missense mutations in SPG30, an autosomal recessive spastic paraplegia: distinct phenotypes according to the nature of the mutations. Eur. J. Hum. Genet. 2012;20:645–649. - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
