

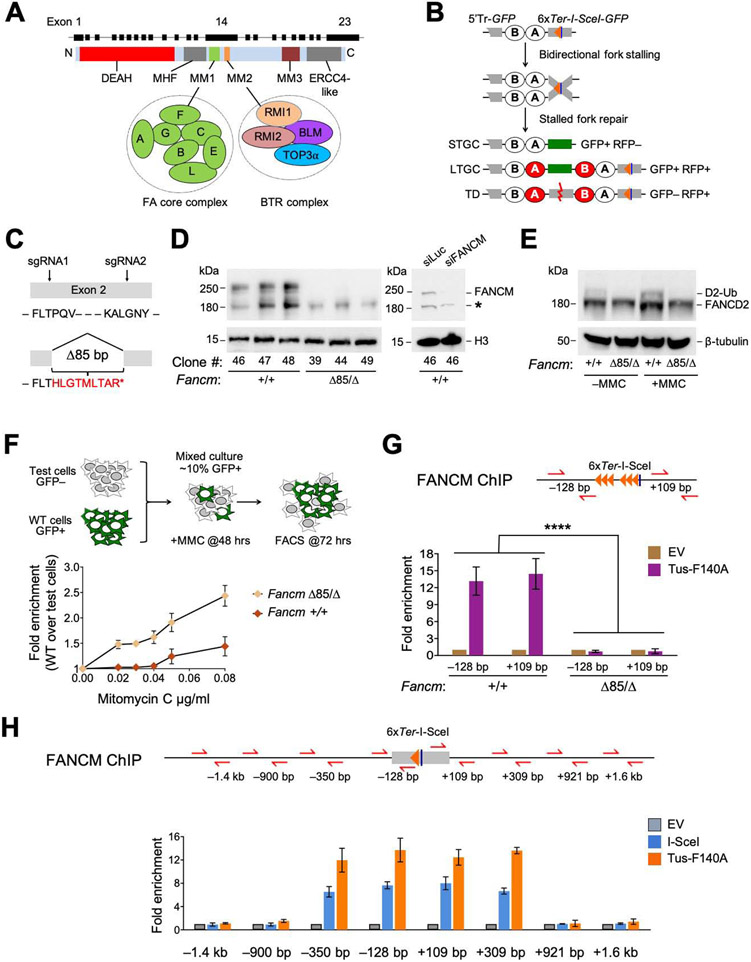
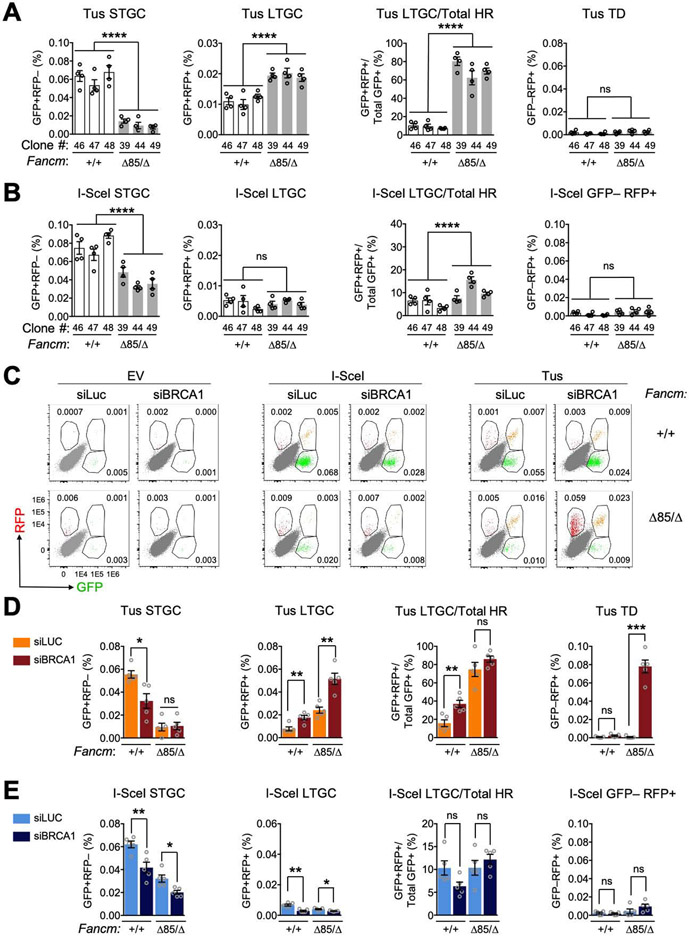
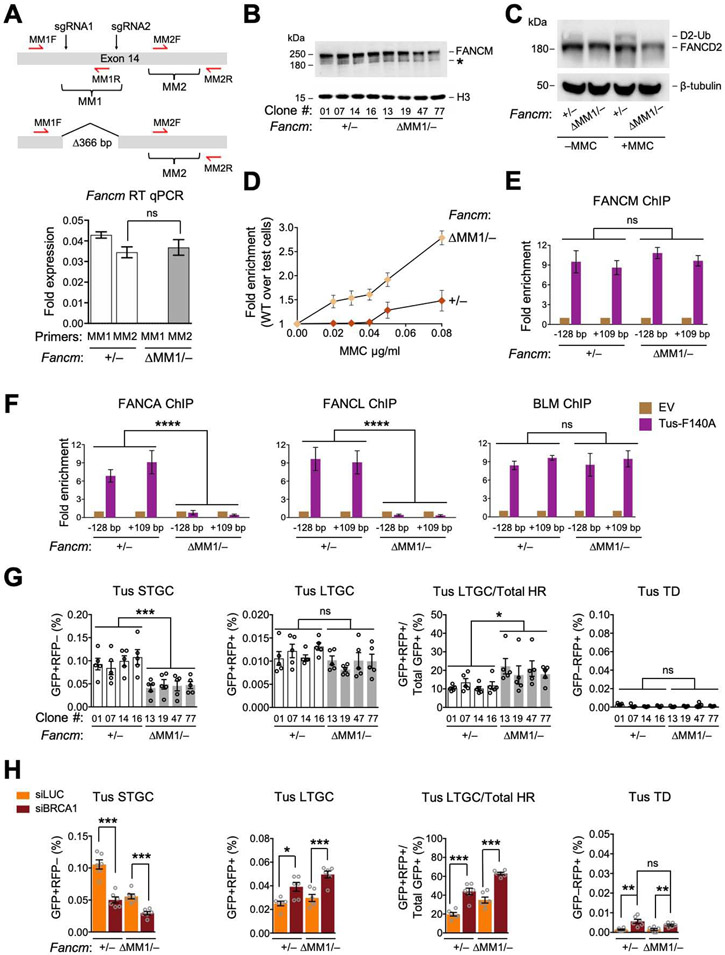
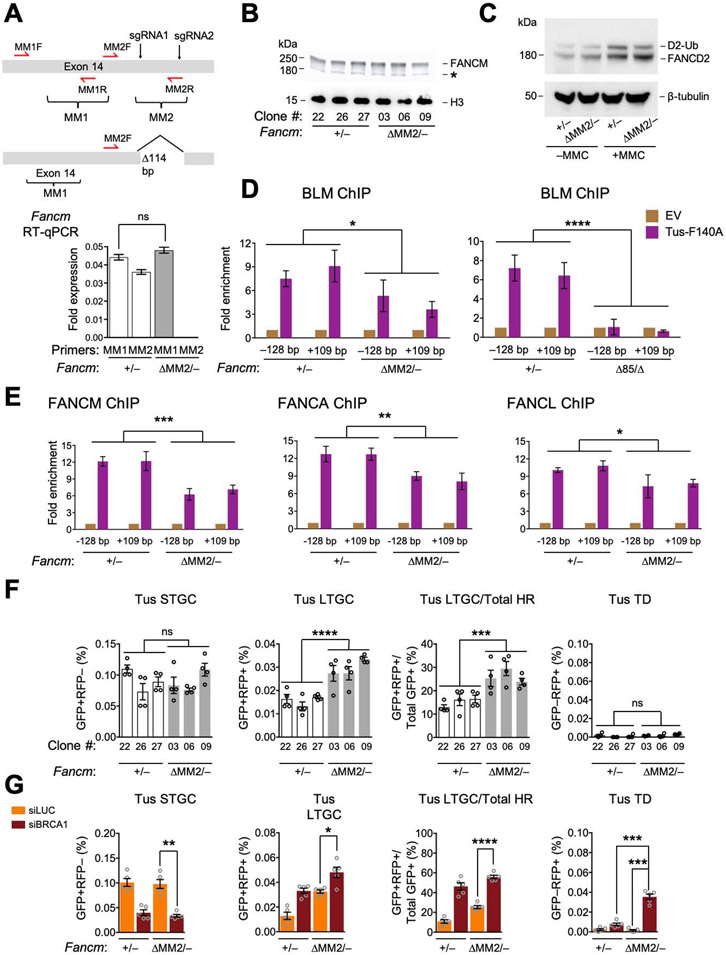
FANCM regulates repair pathway choice at stalled replication forks
- PMID: 33882298
- PMCID: PMC8180084
- DOI: 10.1016/j.molcel.2021.03.044
FANCM regulates repair pathway choice at stalled replication forks
Abstract
Repair pathway "choice" at stalled mammalian replication forks is an important determinant of genome stability; however, the underlying mechanisms are poorly understood. FANCM encodes a multi-domain scaffolding and motor protein that interacts with several distinct repair protein complexes at stalled forks. Here, we use defined mutations engineered within endogenous Fancm in mouse embryonic stem cells to study how Fancm regulates stalled fork repair. We find that distinct FANCM repair functions are enacted by molecularly separable scaffolding domains. These findings define FANCM as a key mediator of repair pathway choice at stalled replication forks and reveal its molecular mechanism. Notably, mutations that inactivate FANCM ATPase function disable all its repair functions and "trap" FANCM at stalled forks. We find that Brca1 hypomorphic mutants are synthetic lethal with Fancm null or Fancm ATPase-defective mutants. The ATPase function of FANCM may therefore represent a promising "druggable" target for therapy of BRCA1-linked cancer.
Keywords: BRCA1; Bloom’s syndrome helicase; FANCM; Fanconi anemia; break-induced replication; genomic instability; homologous recombination; replication restart; synthetic lethality; tandem duplication.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
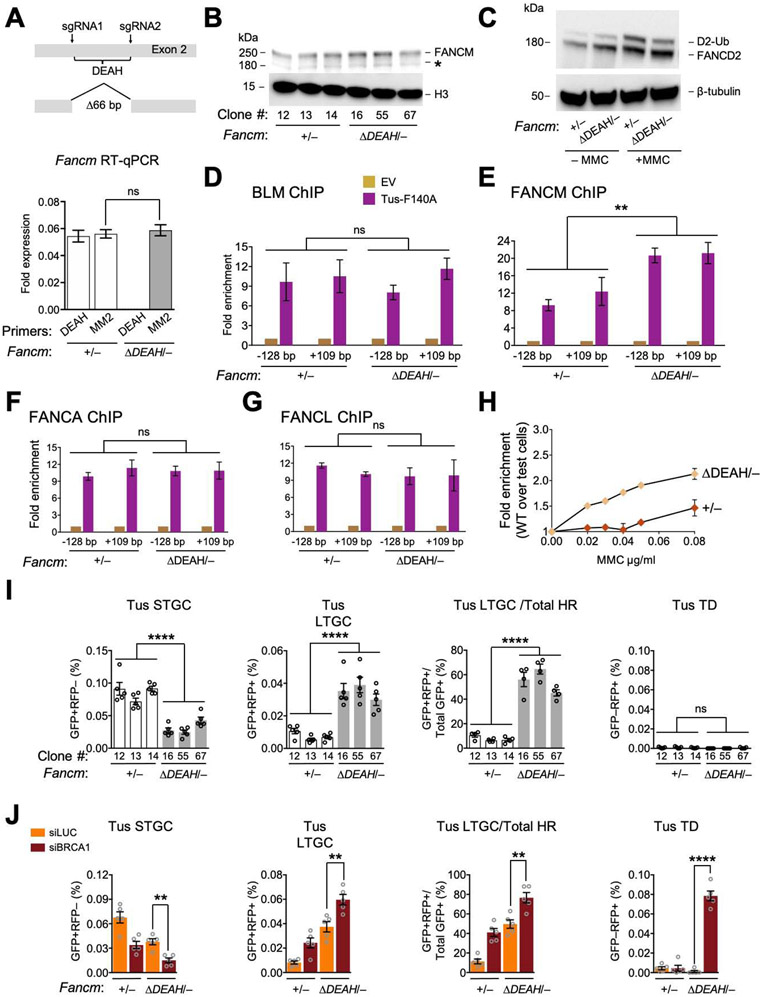

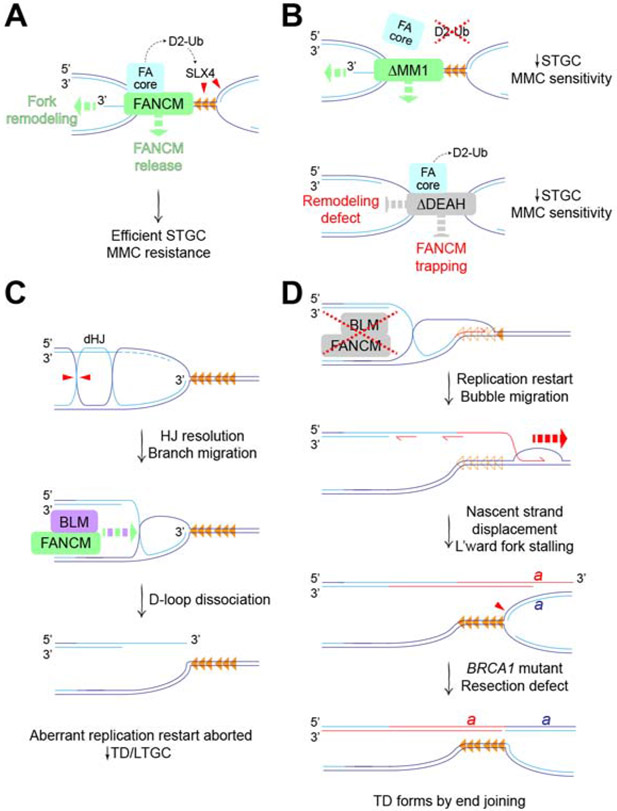
References
-
- Adamo A, Collis SJ, Adelman CA, Silva N, Horejsi Z, Ward JD, Martinez-Perez E, Boulton SJ, and La Volpe A (2010). Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell 39, 25–35. - PubMed
-
- Bakker ST, van de Vrugt HJ, Rooimans MA, Oostra AB, Steltenpool J, Delzenne-Goette E, van der Wal A, van der Valk M, Joenje H, te Riele H, et al. (2009). Fancmdeficient mice reveal unique features of Fanconi anemia complementation group M. Human molecular genetics 18, 3484–3495. - PubMed
-
- Bogliolo M, Bluteau D, Lespinasse J, Pujol R, Vasquez N, d'Enghien CD, Stoppa-Lyonnet D, Leblanc T, Soulier J, and Surrallés J (2018). Biallelic truncating FANCM mutations cause early-onset cancer but not Fanconi anemia. Genetics in medicine : official journal of the American College of Medical Genetics 20, 458–463. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
