

k-mer-Based Metagenomics Tools Provide a Fast and Sensitive Approach for the Detection of Viral Contaminants in Biopharmaceutical and Vaccine Manufacturing Applications Using Next-Generation Sequencing
- PMID: 33883263
- PMCID: PMC8546726
- DOI: 10.1128/mSphere.01336-20
k-mer-Based Metagenomics Tools Provide a Fast and Sensitive Approach for the Detection of Viral Contaminants in Biopharmaceutical and Vaccine Manufacturing Applications Using Next-Generation Sequencing
Abstract
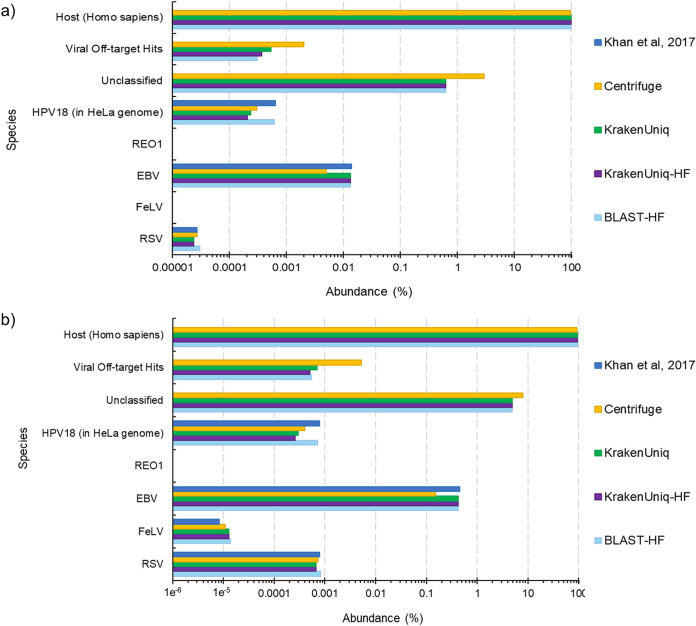
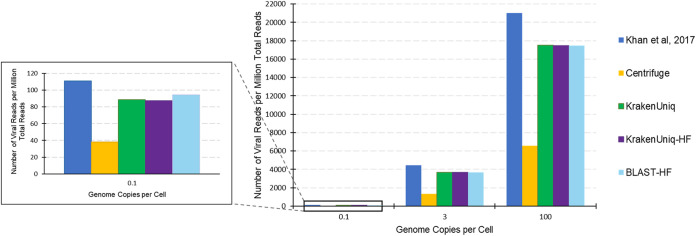
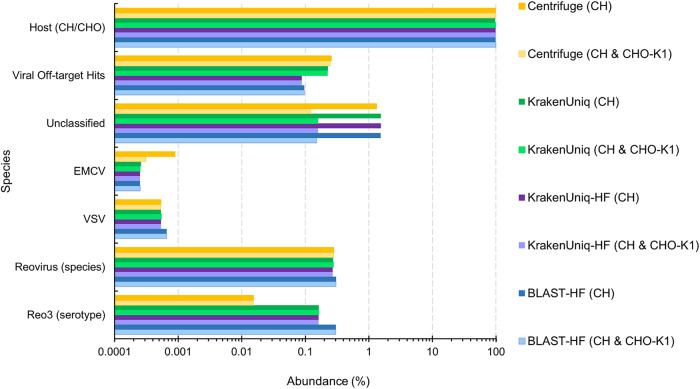
Adventitious agent detection during the production of vaccines and biotechnology-based medicines is of critical importance to ensure the final product is free from any possible viral contamination. Increasing the speed and accuracy of viral detection is beneficial as a means to accelerate development timelines and to ensure patient safety. Here, several rapid viral metagenomics approaches were tested on simulated next-generation sequencing (NGS) data sets and existing data sets from virus spike-in studies done in CHO-K1 and HeLa cell lines. It was observed that these rapid methods had comparable sensitivity to full-read alignment methods used for NGS viral detection for these data sets, but their specificity could be improved. A method that first filters host reads using KrakenUniq and then selects the virus classification tool based on the number of remaining reads is suggested as the preferred approach among those tested to detect nonlatent and nonendogenous viruses. Such an approach shows reasonable sensitivity and specificity for the data sets examined and requires less time and memory as full-read alignment methods.IMPORTANCE Next-generation sequencing (NGS) has been proposed as a complementary method to detect adventitious viruses in the production of biotherapeutics and vaccines to current in vivo and in vitro methods. Before NGS can be established in industry as a main viral detection technology, further investigation into the various aspects of bioinformatics analyses required to identify and classify viral NGS reads is needed. In this study, the ability of rapid metagenomics tools to detect viruses in biopharmaceutical relevant samples is tested and compared to recommend an efficient approach. The results showed that KrakenUniq can quickly and accurately filter host sequences and classify viral reads and had comparable sensitivity and specificity to slower full read alignment approaches, such as BLASTn, for the data sets examined.
Keywords: Chinese hamster ovary cells; HeLa cells; adventitious agent testing; next-generation sequencing; vaccine; viral metagenomics; virus detection.
Copyright © 2021 MacDonald et al.
Figures
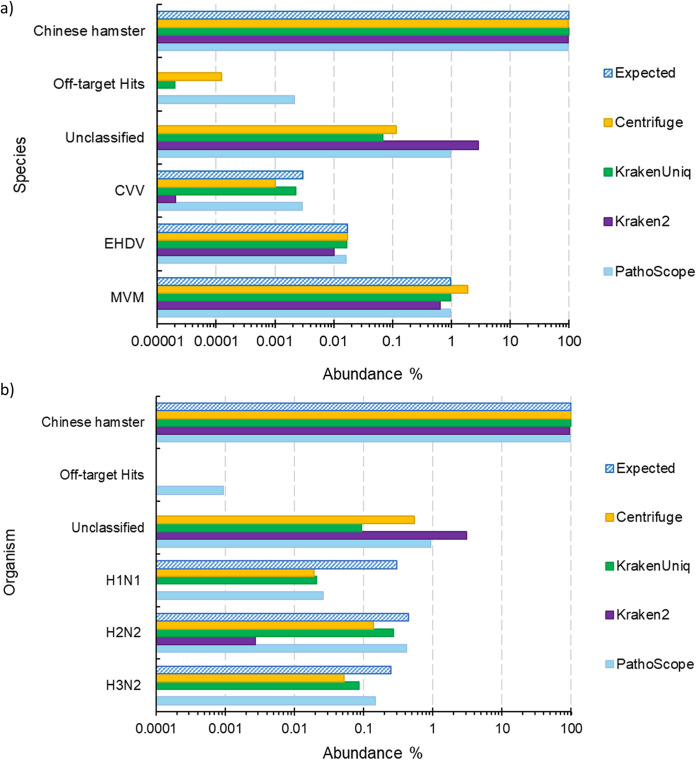
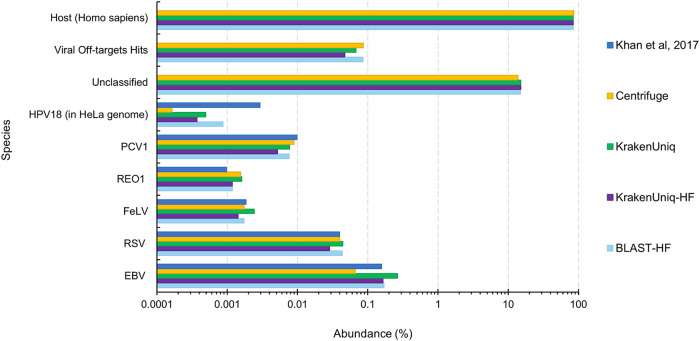
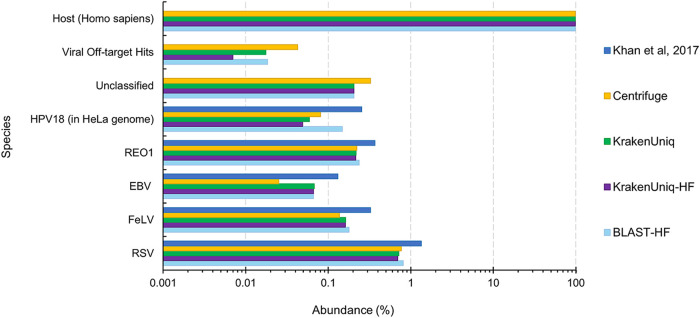
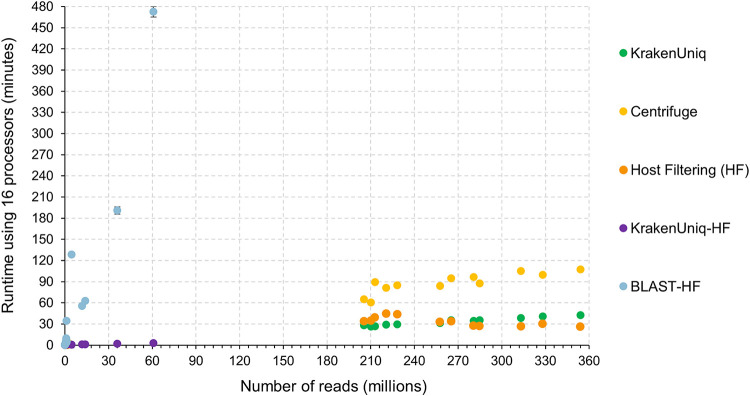

Similar articles
-
Cataloguing the taxonomic origins of sequences from a heterogeneous sample using phylogenomics: applications in adventitious agent detection.PDA J Pharm Sci Technol. 2014 Nov-Dec;68(6):602-18. doi: 10.5731/pdajpst.2014.01023. PDA J Pharm Sci Technol. 2014. PMID: 25475635
-
Detection of adventitious agents using next-generation sequencing.PDA J Pharm Sci Technol. 2014 Nov-Dec;68(6):651-60. doi: 10.5731/pdajpst.2014.01025. PDA J Pharm Sci Technol. 2014. PMID: 25475640
-
Validation of a Next Generation Sequencing Method for adventitious agents detection in a live vaccine matrix.Biologicals. 2025 May;90:101828. doi: 10.1016/j.biologicals.2025.101828. Epub 2025 Apr 2. Biologicals. 2025. PMID: 40179635 Review.
-
Report of the second international conference on next generation sequencing for adventitious virus detection in biologics for humans and animals.Biologicals. 2020 Sep;67:94-111. doi: 10.1016/j.biologicals.2020.06.002. Epub 2020 Jul 11. Biologicals. 2020. PMID: 32660862 Free PMC article.
-
Need for new technologies for detection of adventitious agents in vaccines and other biological products.PDA J Pharm Sci Technol. 2014 Nov-Dec;68(6):556-62. doi: 10.5731/pdajpst.2014.01012. PDA J Pharm Sci Technol. 2014. PMID: 25475629 Review.
Cited by
-
Next-Generation Sequencing for the Detection of Microbial Agents in Avian Clinical Samples.Vet Sci. 2023 Dec 4;10(12):690. doi: 10.3390/vetsci10120690. Vet Sci. 2023. PMID: 38133241 Free PMC article. Review.
-
INSaFLU-TELEVIR: an open web-based bioinformatics suite for viral metagenomic detection and routine genomic surveillance.Genome Med. 2024 Apr 25;16(1):61. doi: 10.1186/s13073-024-01334-3. Genome Med. 2024. PMID: 38659008 Free PMC article.
-
Viral Integration Plays a Minor Role in the Development and Prognostication of Oral Squamous Cell Carcinoma.Cancers (Basel). 2022 Oct 24;14(21):5213. doi: 10.3390/cancers14215213. Cancers (Basel). 2022. PMID: 36358632 Free PMC article.
References
-
- Barone PW, Wiebe ME, Leung JC, Hussein ITM, Keumurian FJ, Bouressa J, Brussel A, Chen D, Chong M, Dehghani H, Gerentes L, Gilbert J, Gold D, Kiss R, Kreil TR, Labatut R, Li Y, Müllberg J, Mallet L, Menzel C, Moody M, Monpoeho S, Murphy M, Plavsic M, Roth N, Roush D, Ruffing M, Schicho R, Snyder R, Stark D, Zhang C, Wolfrum J, Sinskey AJ, Spring SL. 2020. Viral contamination in biologic manufacture and implications for emerging therapies. Nat Biotechnol 38:563–572. doi: 10.1038/s41587-020-0507-2. - DOI - PubMed
-
- Garnick RL. 1996. Experience with viral contamination in cell culture. Dev Biol Stand 88:49–56. - PubMed
-
- Nims RW. 2006. Detection of adventitious viruses in biologicals-a rare occurrence. Dev Biol (Basel) 123:153–164. - PubMed
-
- Bethencourt V. 2009. Virus stalls Genzyme plant. Nat Biotechnol 27:681–681. doi: 10.1038/nbt0809-681a. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
