

PD-L1 tumor-intrinsic signaling and its therapeutic implication in triple-negative breast cancer
- PMID: 33884962
- PMCID: PMC8119208
- DOI: 10.1172/jci.insight.131458
PD-L1 tumor-intrinsic signaling and its therapeutic implication in triple-negative breast cancer
Abstract
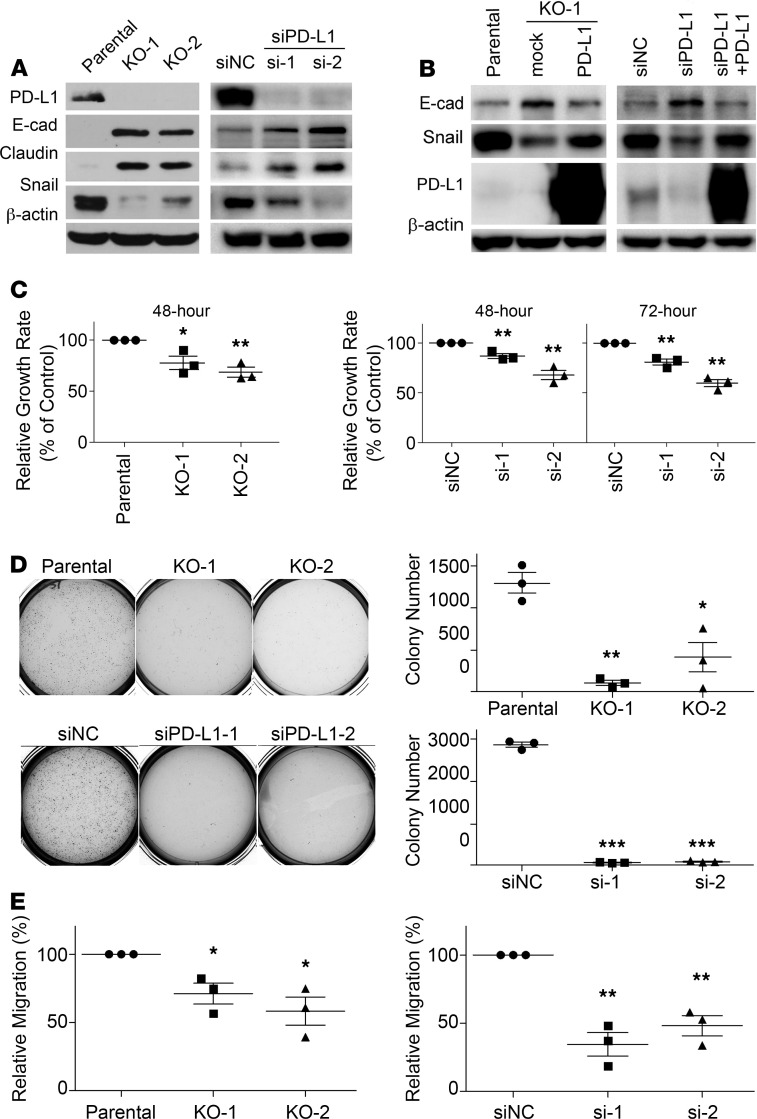
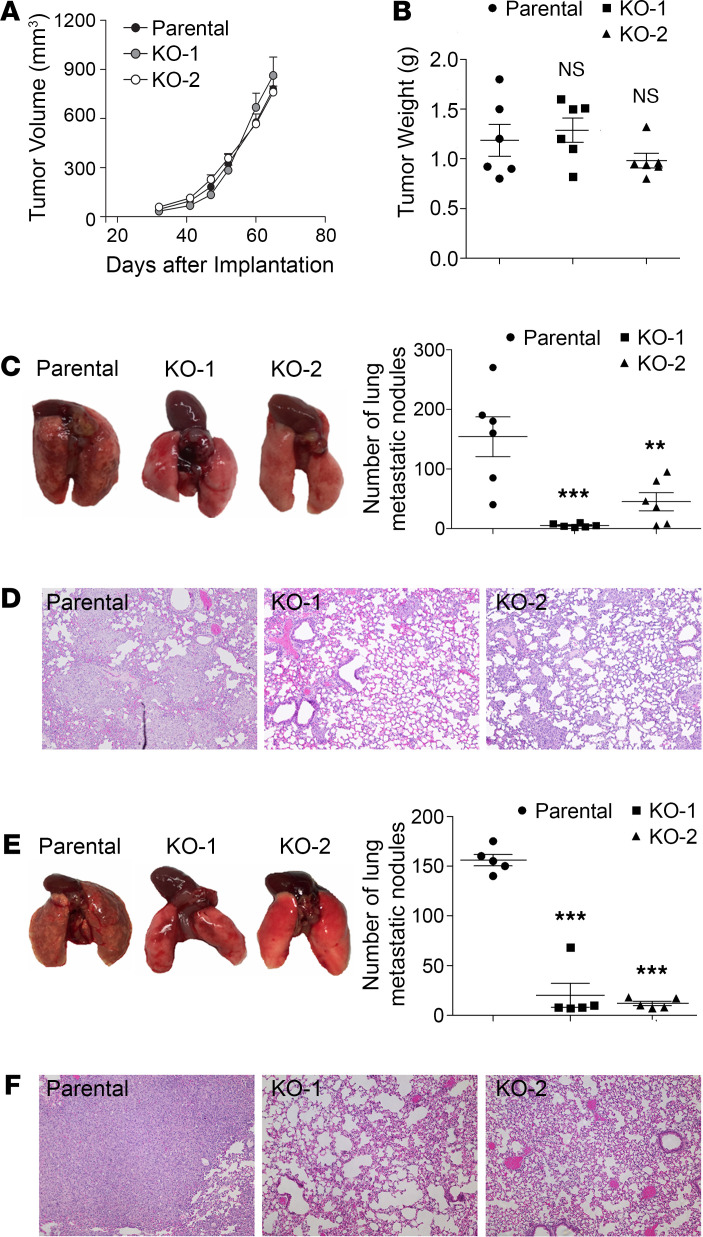
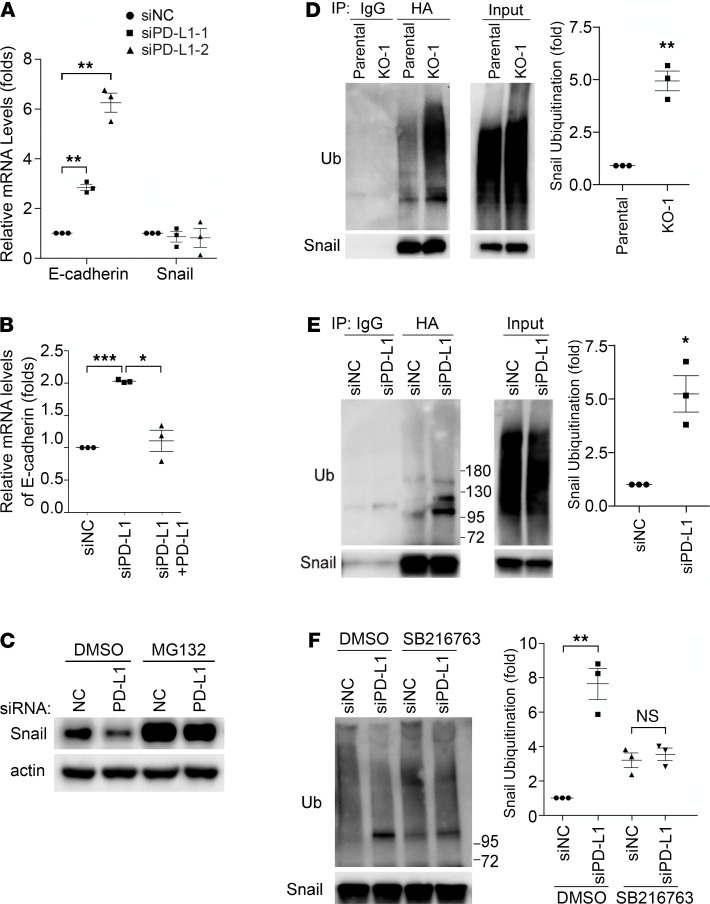
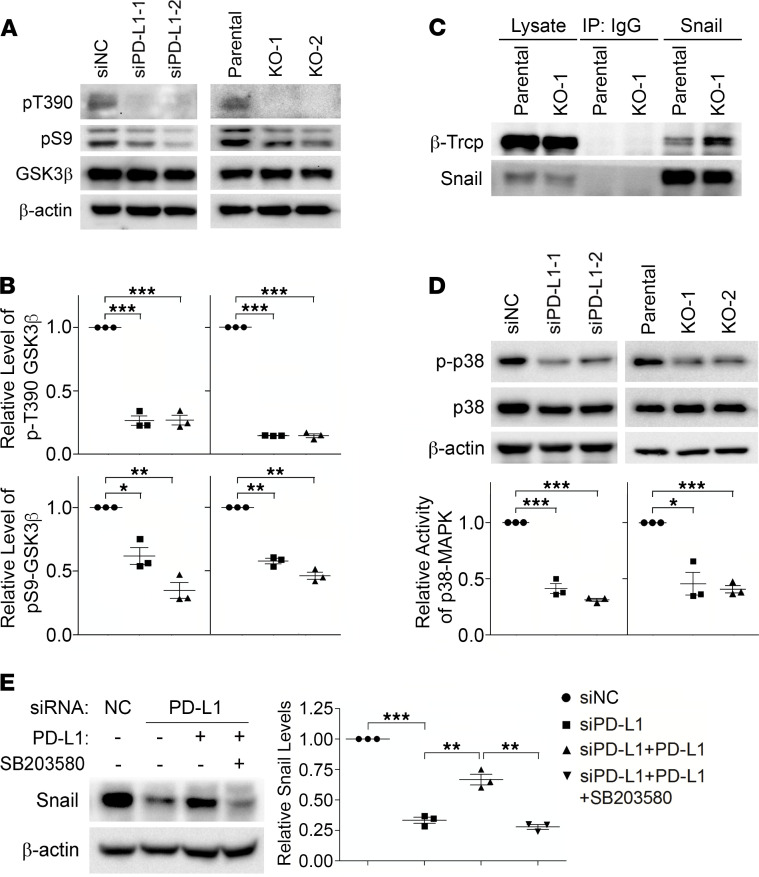
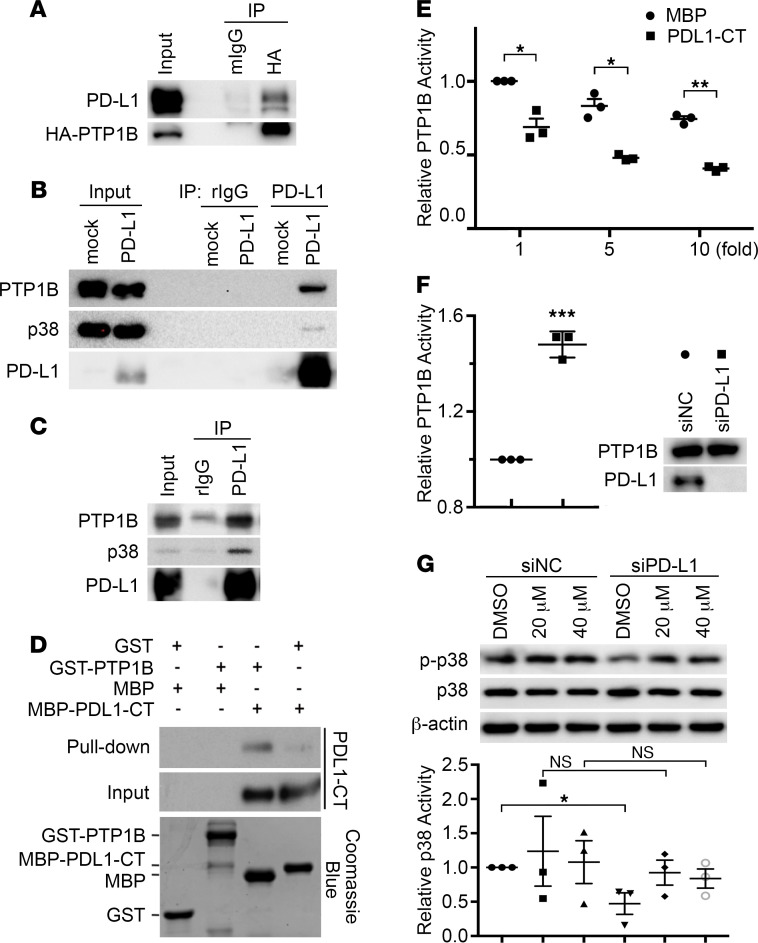
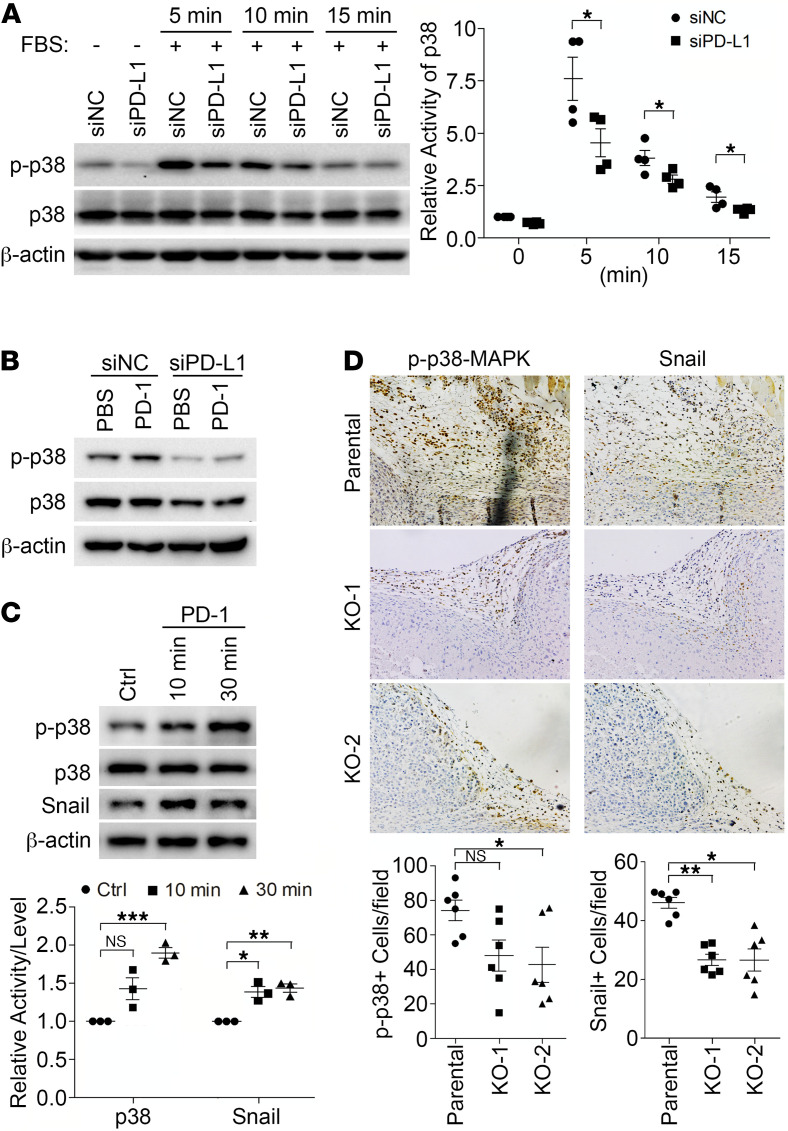
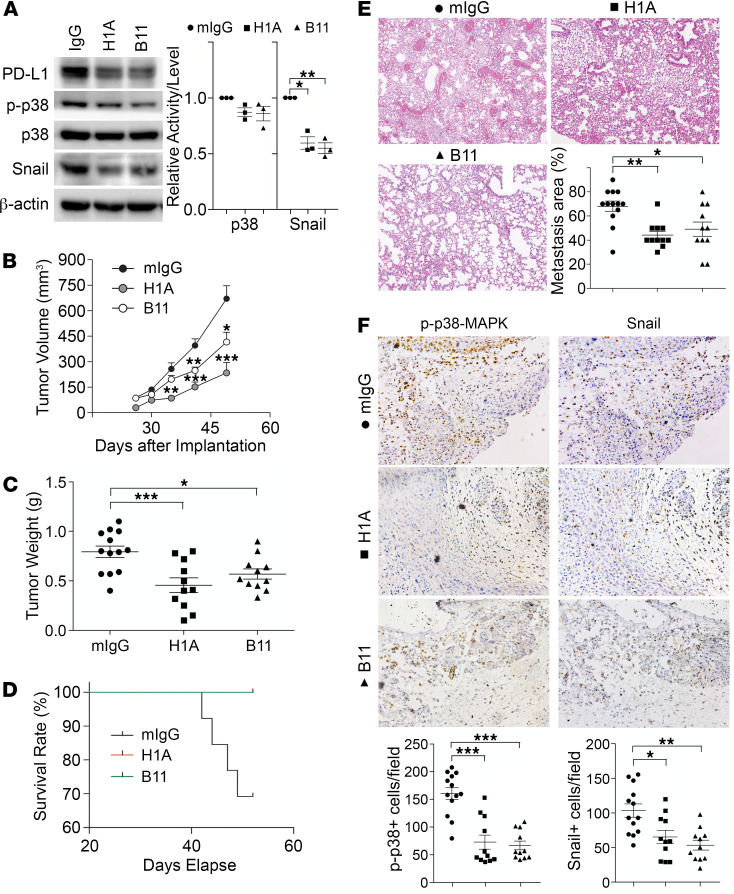
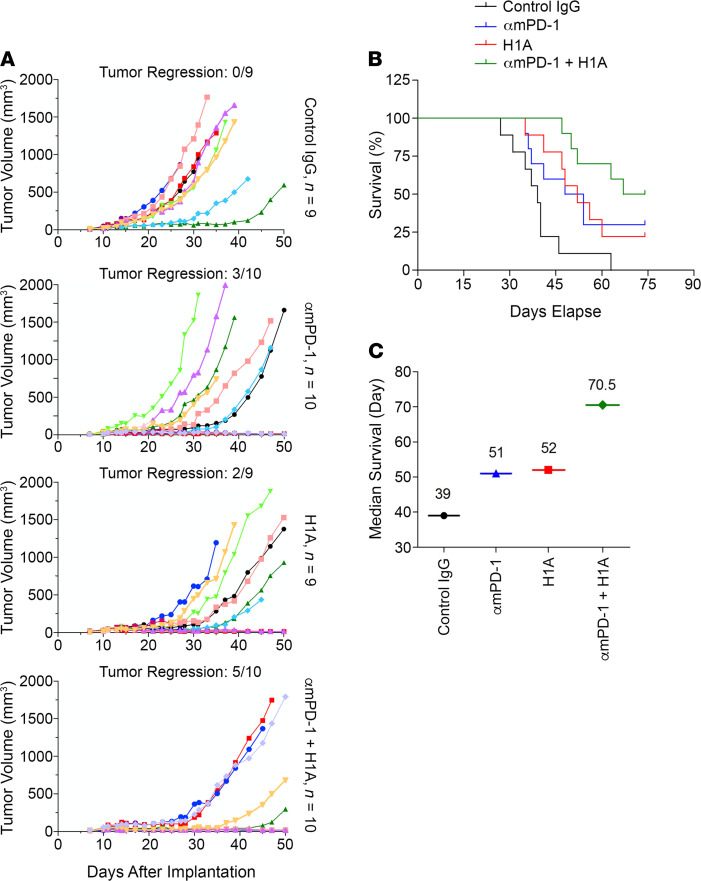
Although the immune checkpoint role of programmed death ligand 1 (PD-L1) has been established and targeted in cancer immunotherapy, the tumor-intrinsic role of PD-L1 is less appreciated in tumor biology and therapeutics development, partly because of the incomplete mechanistic understanding. Here we demonstrate a potentially novel mechanism by which PD-L1 promotes the epithelial-mesenchymal transition (EMT) in triple-negative breast cancer (TNBC) cells by suppressing the destruction of the EMT transcription factor Snail. PD-L1 directly binds to and inhibits the tyrosine phosphatase PTP1B, thus preserving p38-MAPK activity that phosphorylates and inhibits glycogen synthase kinase 3β (GSK3β). Via this mechanism, PD-L1 prevents the GSK3β-mediated phosphorylation, ubiquitination, and degradation of Snail and consequently promotes the EMT and metastatic potential of TNBC. Significantly, PD-L1 antibodies that confine the tumor-intrinsic PD-L1/Snail pathway restricted TNBC progression in immunodeficient mice. More importantly, targeting both tumor-intrinsic and tumor-extrinsic functions of PD-L1 showed strong synergistic tumor suppression effect in an immunocompetent TNBC mouse model. Our findings support that PD-L1 intrinsically facilitates TNBC progression by promoting the EMT, and this potentially novel PD-L1 signaling pathway could be targeted for better clinical management of PD-L1-overexpressing TNBCs.
Keywords: Breast cancer; Immunotherapy; Oncology; Therapeutics; Ubiquitin-proteosome system.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
