

Pharmacophore model, docking, QSAR, and molecular dynamics simulation studies of substituted cyclic imides and herbal medicines as COX-2 inhibitors
- PMID: 33889764
- PMCID: PMC8047494
- DOI: 10.1016/j.heliyon.2021.e06605
Pharmacophore model, docking, QSAR, and molecular dynamics simulation studies of substituted cyclic imides and herbal medicines as COX-2 inhibitors
Abstract
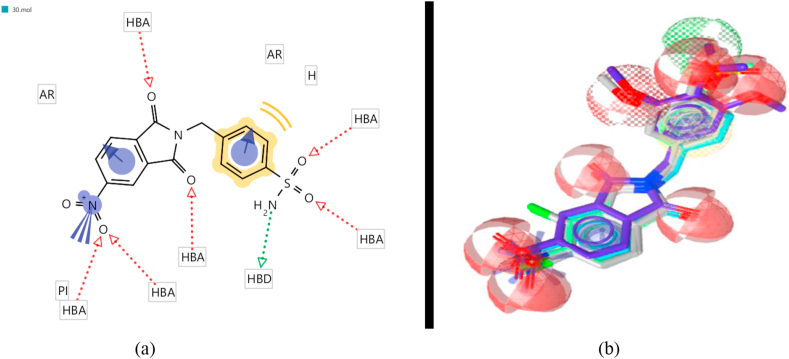
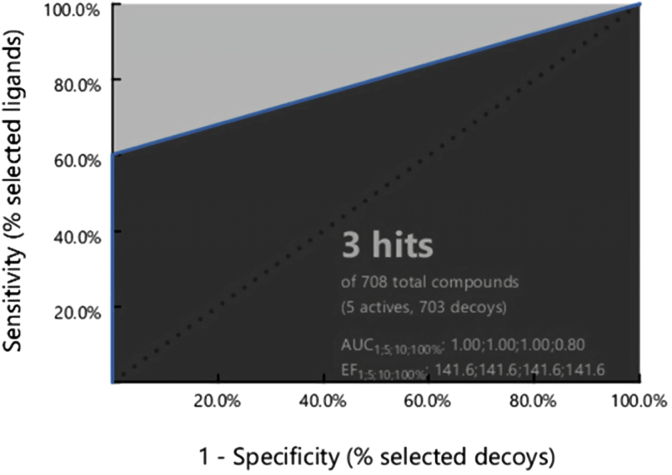
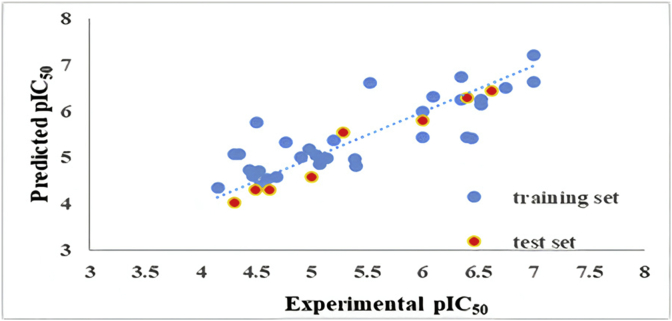
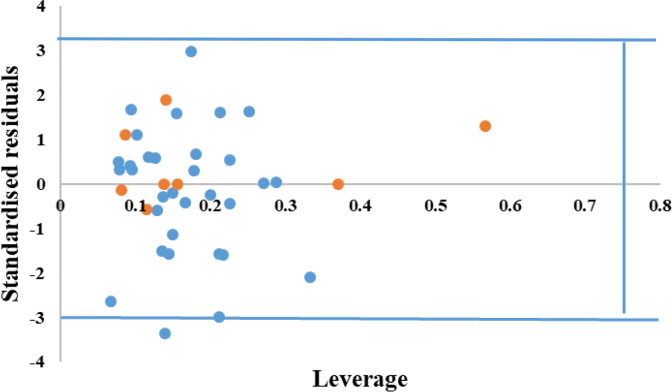
Cyclooxygenase-2 (COX-2) enzyme inhibitors have not eliminated the necessity for developed drugs not only in the nonsteroidal anti-inflammatory drug (NSAIDs) area, but also in other therapeutic applications including prevention of cancer and Alzheimer's disease. A series of novel substituted cyclic imides have been reported as selective COX-2 inhibitors. To understand the structural features responsible for their activity, a 3D validated pharmacophore and quantitative structure-activity relationship (QSAR) model have been developed. The values of enrichment factor (EF), goodness of hit score (GH), area under the ROC curve (AUC), sensitivity, and specificity refer to the good ability of the pharmacophore model to identify active compounds. Multiple linear regression (MLR) produced statistically significant QSAR model with (R2 training = 0.763, R2 test = 0.96) and predictability (Q2 training = 0.66, Q2 test = 0.84). Then, using the pharmacophore and QSAR models, eight authenticated botanicals in two herbal medicines and the ZINC compounds database, were virtually screened for ligands to COX-2. The retrieved hits which also obey lipinski's rule of five (RO5) were docked in the COX-2 3D structure to investigate their binding mode and affinity. Finally, based on the docking results, nine molecules were prioritized as promising hits that could be used as leads to discover novel COX-2 inhibitors. COX-2 inhibition of most of these hits has not been reported previously. Ten-nanosecond molecular dynamics simulation (10-ns MD) was performed on the initial structure COX-2 complex with ZINC000113253375 and ZINC000043170560 resulted from the docking. Our utilization of the 3D pharmacophore model, QSAR, molecular docking, and molecular dynamics simulation trials can be a potent strategy to successfully predict activity, efficiently design drugs, and screen large numbers of new compounds as active drug candidates.
Keywords: Cyclooxygenase-2; Docking; Molecular dynamics simulation; Pharmacophore; QSAR; Virtual screening.
© 2021 The Author(s).
Conflict of interest statement
The authors declare no conflict of interest.
Figures
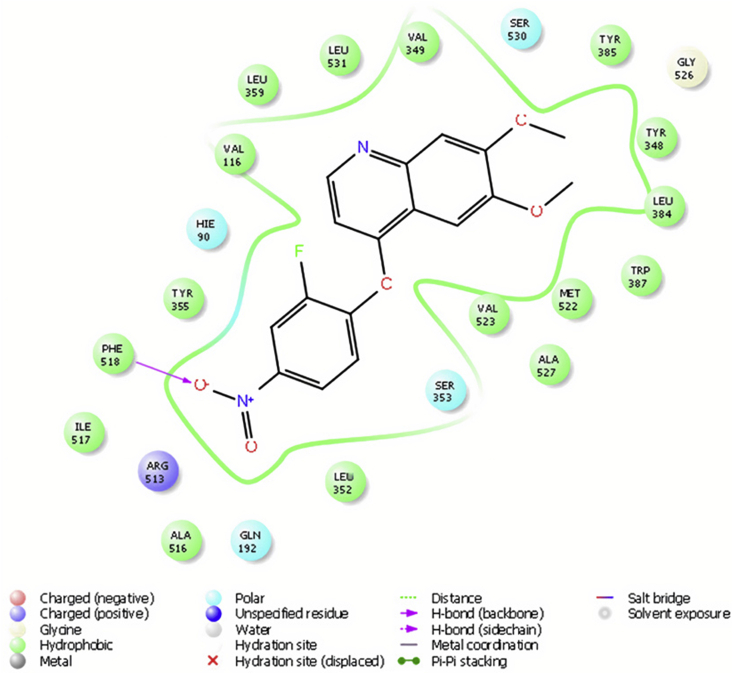
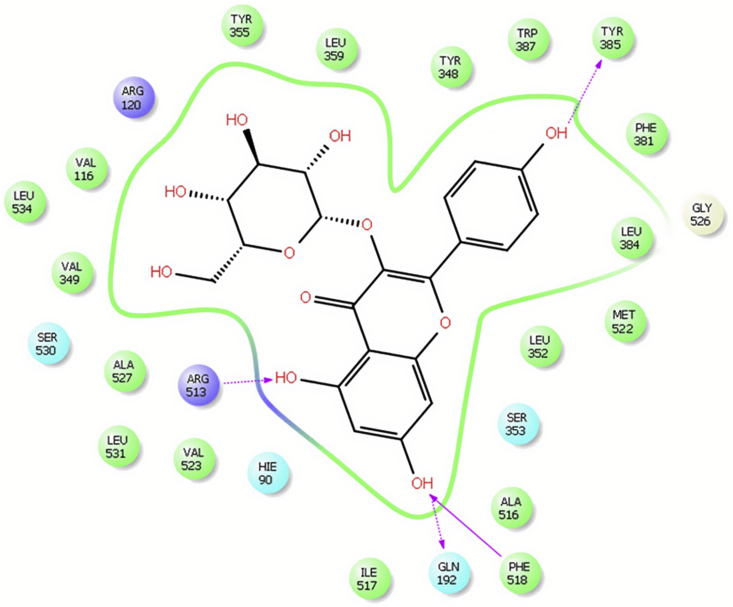
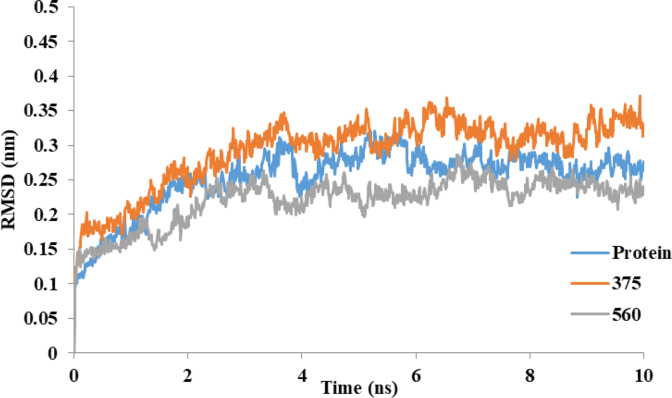
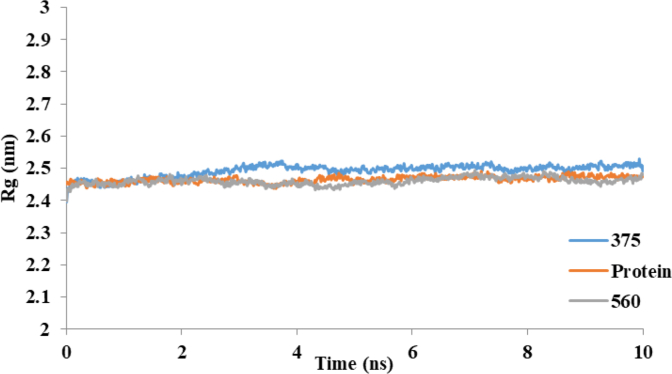
References
-
- Abouzid K., Frohberg P., Lehmann J., Decker M. 6-Aryl-4-oxohexanoic acids: synthesis, effects on eicosanoid biosynthesis, and anti-inflammatory in vivo-activities. Med. Chem. 2007;3:433–438. - PubMed
-
- Ulbrich H., Fiebich B., Dannhardt G. Cyclooxygenase-1/2 (COX-1/COX-2) and 5-lipoxygenase (5-LOX) inhibitors of the 6,7-diaryl-2,3-1H-dihydropyrrolizine type. ChemInform. 2003;34(20):953–959. - PubMed
-
- Dannhardt G., Laufer S. Structural approaches to explain the selectivity of COX-2 inhibitors: is there a common pharmacophore? Curr. Med. Chem. 2000;7(11):1101–1112. - PubMed
-
- Kim H.-J., Chae C.H., Yi K.Y., Park K.-L., Yoo S. Computational studies of COX-2 inhibitors: 3D-QSAR and docking. Bioorg. Med. Chem. 2004;12(7):1629–1641. - PubMed
-
- Almansa C., de Arriba A.F., Cavalcanti F.L., Gómez L.A., Miralles A., Merlos M., Forn J. Synthesis and SAR of a new series of COX-2-selective inhibitors: pyrazolo [1,5-a] pyrimidines. J. Med. Chem. 2001;44(3):350–361. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
