

Metabolic-associated fatty liver disease and lipoprotein metabolism
- PMID: 33892169
- PMCID: PMC8324684
- DOI: 10.1016/j.molmet.2021.101238
Metabolic-associated fatty liver disease and lipoprotein metabolism
Abstract
Background: Non-alcoholic fatty liver disease, or as recently proposed 'metabolic-associated fatty liver disease' (MAFLD), is characterized by pathological accumulation of triglycerides and other lipids in hepatocytes. This common disease can progress from simple steatosis to steatohepatitis, and eventually end-stage liver diseases. MAFLD is closely related to disturbances in systemic energy metabolism, including insulin resistance and atherogenic dyslipidemia.
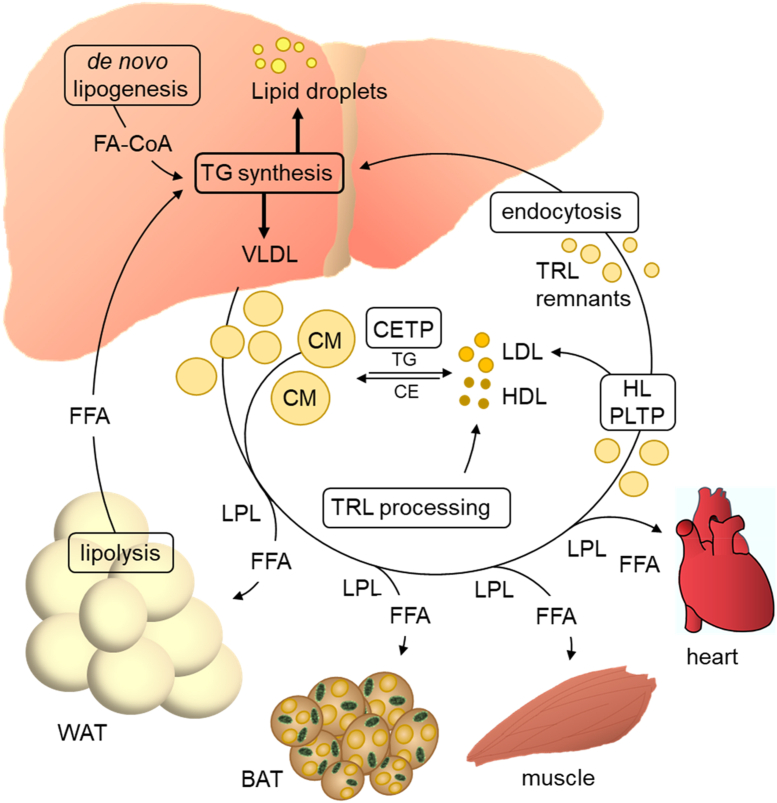
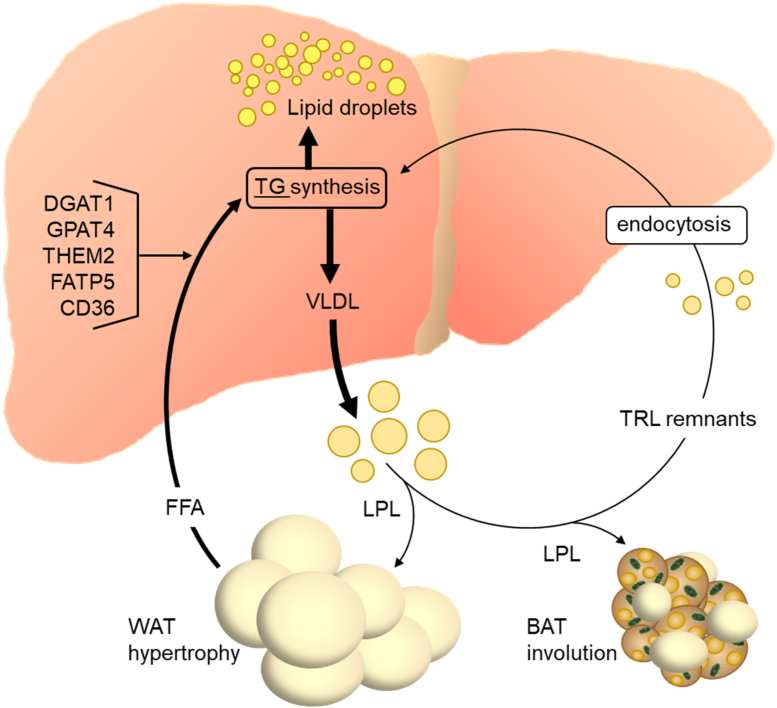
Scope of review: The liver is the central organ in lipid metabolism by secreting very low density lipoproteins (VLDL) and, on the other hand, by internalizing fatty acids and lipoproteins. This review article discusses recent research addressing hepatic lipid synthesis, VLDL production, and lipoprotein internalization as well as the lipid exchange between adipose tissue and the liver in the context of MAFLD.
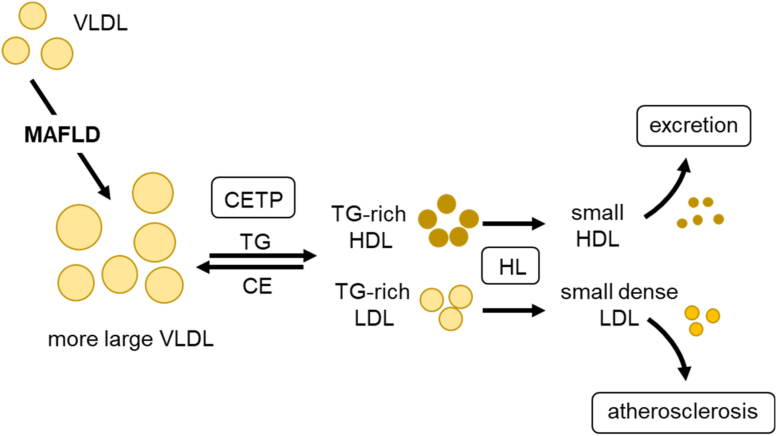
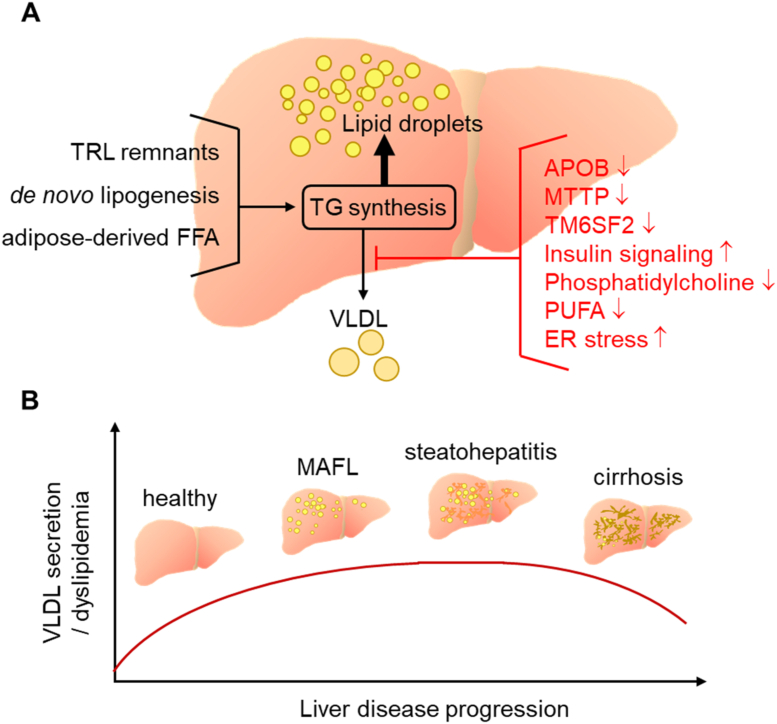
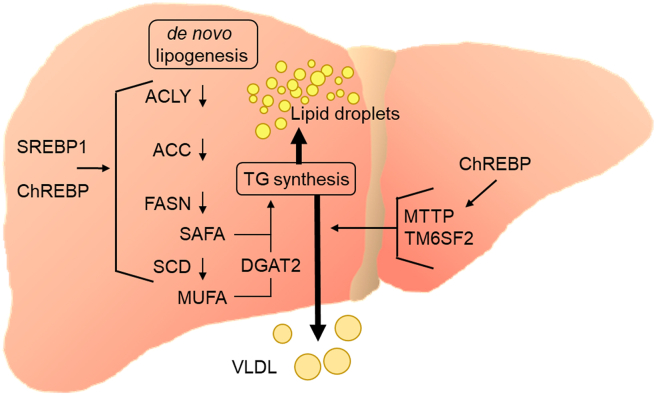
Major conclusions: Liver steatosis in MAFLD is triggered by excessive hepatic triglyceride synthesis utilizing fatty acids derived from white adipose tissue (WAT), de novo lipogenesis (DNL) and endocytosed remnants of triglyceride-rich lipoproteins. In consequence of high hepatic lipid content, VLDL secretion is enhanced, which is the primary cause of complex dyslipidemia typical for subjects with MAFLD. Interventions reducing VLDL secretory capacity attenuate dyslipidemia while they exacerbate MAFLD, indicating that the balance of lipid storage versus secretion in hepatocytes is a critical parameter determining disease outcome. Proof of concept studies have shown that promoting lipid storage and energy combustion in adipose tissues reduces hepatic lipid load and thus ameliorates MAFLD. Moreover, hepatocellular triglyceride synthesis from DNL and WAT-derived fatty acids can be targeted to treat MAFLD. However, more research is needed to understand how individual transporters, enzymes, and their isoforms affect steatosis and dyslipidemia in vivo, and whether these two aspects of MAFLD can be selectively treated. Processing of cholesterol-enriched lipoproteins appears less important for steatosis. It may, however, modulate inflammation and consequently MAFLD progression.
Keywords: Adipose tissue; De novo lipogenesis; Lipoprotein; Liver; NAFLD; Triglycerides.
Copyright © 2021 The Authors. Published by Elsevier GmbH.. All rights reserved.
Figures
References
-
- Dash S., Xiao C., Morgantini C., Lewis G.F. New insights into the regulation of chylomicron production. Annual Review of Nutrition. 2015;35:265–294. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
