

Mitochondrial analysis of oribatid mites provides insights into their atypical tRNA annotation, genome rearrangement and evolution
- PMID: 33892790
- PMCID: PMC8063316
- DOI: 10.1186/s13071-021-04719-0
Mitochondrial analysis of oribatid mites provides insights into their atypical tRNA annotation, genome rearrangement and evolution
Abstract
Background: The mitochondrial (mt) genomes of Sarcoptiformes mites typically contain 37 genes. Although the loss of genes is rare in Sarcoptiformes mite mitogenomes, two of the six previously reported oribatid mites (Acariforms: Sarcoptiformes) are reported to have lost parts of their tRNA genes. To confirm whether the tRNA genes were indeed lost and whether the loss is universal, we re-annotated the available oribatid mite sequences and sequenced the mitogenome of Oribatula sakamorii.
Methods: The mitogenome of O. sakamorii was sequenced using an Illumina HiSeq sequencer. The mt tRNA gene was annotated using multi-software combined with a manual annotation approach. Phylogenetic analyses were performed using the maximum likelihood and Bayesian inference methods with concatenated nucleotide and amino acid sequences.
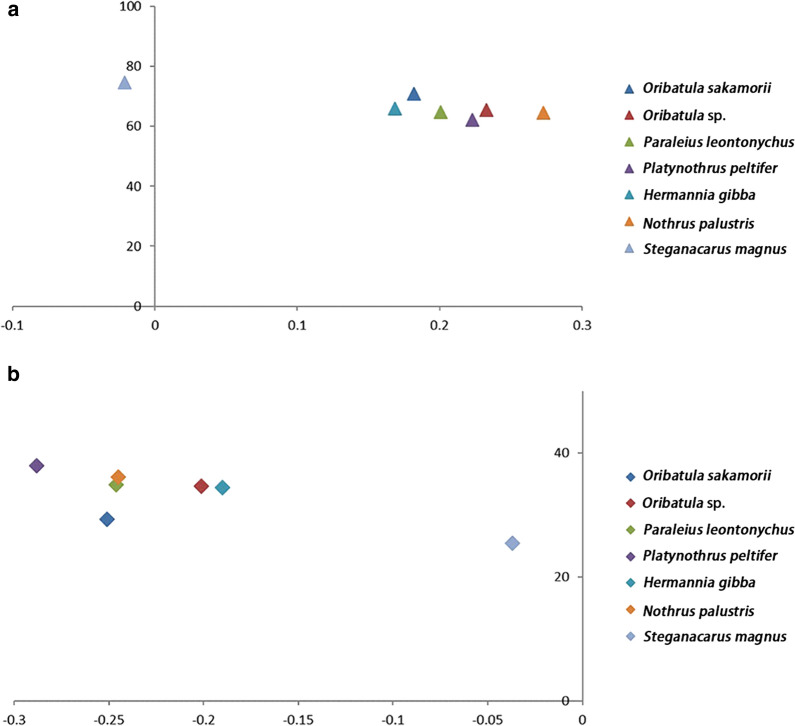
Results: The mitogenomes of O. sakamorii contained 37 genes, including 22 tRNA genes. We identified all mt tRNA genes that were reported as "lost" in Steganacarus magnus and Paraleius leontonychus and revealed certain atypical tRNA annotation errors in oribatid mite sequences. Oribatid mite mitogenomes are characterized by low rates of genetic rearrangement, with six or seven gene blocks conserved between the mitogenome of all species and that of ancestral arthropods. Considering the relative order of the major genes (protein-coding genes and rRNAs), only one or two genes were rearranged with respect to their positions in the ancestral genome. We explored the phylogenetic relationships among the available oribatid mites, and the results confirmed the systematic position of Hermannia in the Crotonioidea superfamily. This was also supported by the synapomorphic gene-derived boundaries.
Conclusions: The tRNA "lost" phenomenon is not universal in oribatid mites. Rather, highly atypical secondary structure of the inferred mt tRNA genes made them unidentifiable using a single type of tRNA search program. The use of multi-software combined with a manual annotation approach can improve the accuracy of tRNA gene annotation. In addition, we identified the precise systematic position of Hermannia and validated that Astigmata is nested in Oribatida.
Keywords: Mitochondrial genome; Oribatid mites; Phylogeny; TRNA re-annotation.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures







References
-
- Zhang ZQ. Animal biodiversity: An outline of higher-level classification and survey of taxonomic richness. New Zealand: Magnolia Press; 2011. - PubMed
-
- Lindquist EE, Krantz GW, Walter DE. A manual of acarology. Lubbock: Texas Tech University Press; 2009.
-
- Oudemans AC. Studie over de sedert 1877 ontworpen systemen der Acari; nieuwe classificatie; phylogenetische beschouwingen. Tijdschr Entomol. 1923;66:49–85.
-
- Baker EW, Crabill RE, Nunes G. Guide to the families of mites. Southwest Nat. 1958;3:238. doi: 10.2307/3669079. - DOI
-
- Woolley TA. A review of the phylogeny of mites. Annu Rev Entomol. 1961;6:263–284. doi: 10.1146/annurev.en.06.010161.001403. - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
