

Disrupting tumor onset and growth via selective cell tagging (SeCT) therapy
- PMID: 33893089
- PMCID: PMC8064634
- DOI: 10.1126/sciadv.abg4038
Disrupting tumor onset and growth via selective cell tagging (SeCT) therapy
Abstract
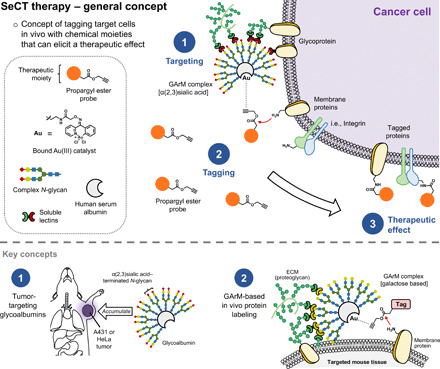
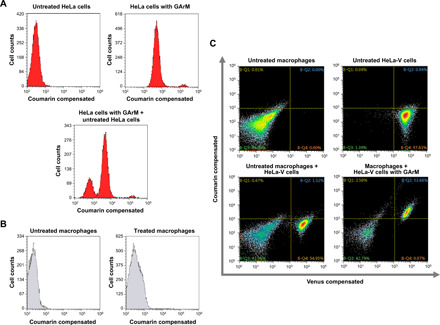
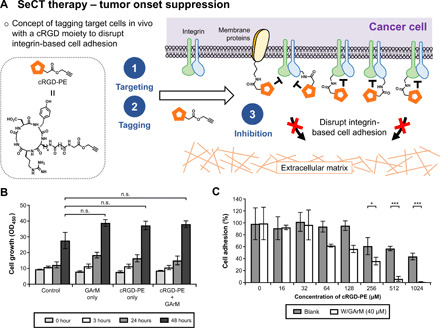
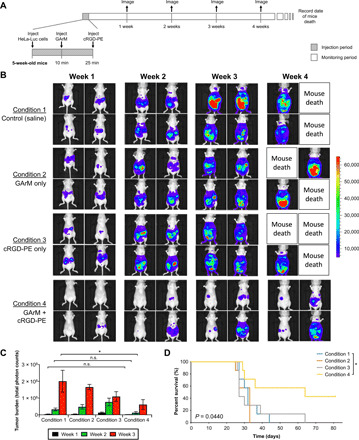
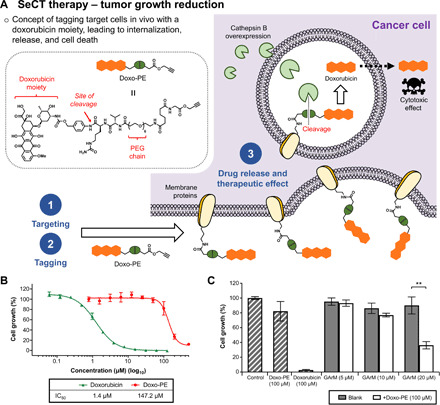
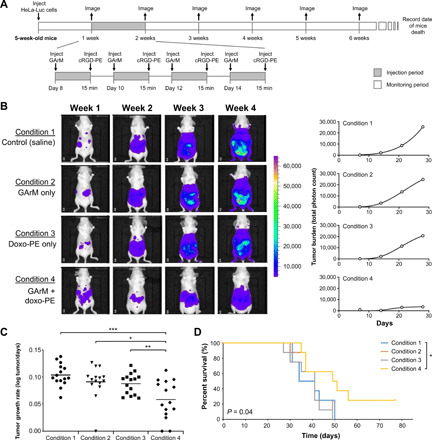
This study presents the early framework of selective cell tagging (SeCT) therapy, which is the concept of preferentially labeling specific cells in vivo with chemical moieties that can elicit a therapeutic response. Using glycosylated artificial metalloenzyme (GArM)-based protein labeling, this study reports two separate functional strategies. In one approach, early tumor onset can be suppressed by tagging cancer cells in living mice with an integrin-blocking cyclic-Arg-Gly-Asp (cRGD) moiety, thereby disrupting cell adhesion onto the extracellular matrix. In another approach, tumor growth in mice can be reduced by tagging with a cytotoxic doxorubicin moiety. Subsequent cell death occurs following internalization and drug release. Overall, experiments have shown that mouse populations receiving the mixture of SeCT labeling reagents exhibited a significant delay/reduction in tumor onset and growth compared with controls. Highlighting its adaptability, this work represents a foundational step for further development of SeCT therapy and its potential therapeutic applications.
Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC).
Figures
References
-
- Tsukiji S., Miyagawa M., Takaoka Y., Tamura T., Hamachi I., Ligand-directed tosyl chemistry for protein labeling in vivo. Nat. Chem. Biol. 5, 341–343 (2009). - PubMed
-
- Tamura T., Kioi Y., Miki T., Tsukiji S., Hamachi I., Fluorophore labeling of native FKBP12 by ligand-directed tosyl chemistry allows detection of its molecular interactions in vitro and in living cells. J. Am. Chem. Soc. 135, 6782–6785 (2013). - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
