

The impact of non-additive genetic associations on age-related complex diseases
- PMID: 33893285
- PMCID: PMC8065056
- DOI: 10.1038/s41467-021-21952-4
The impact of non-additive genetic associations on age-related complex diseases
Abstract
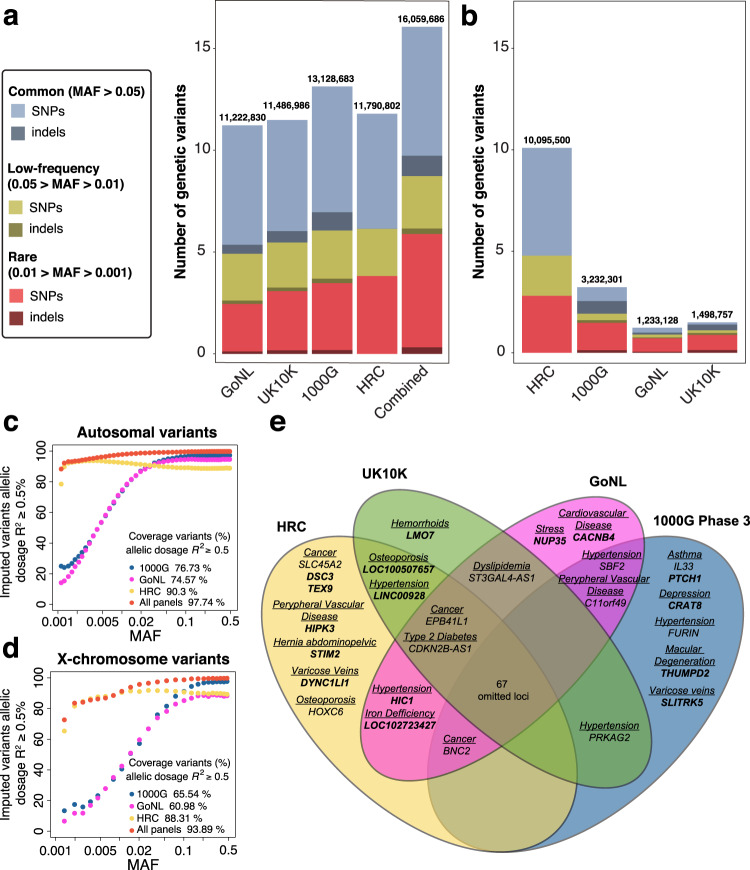
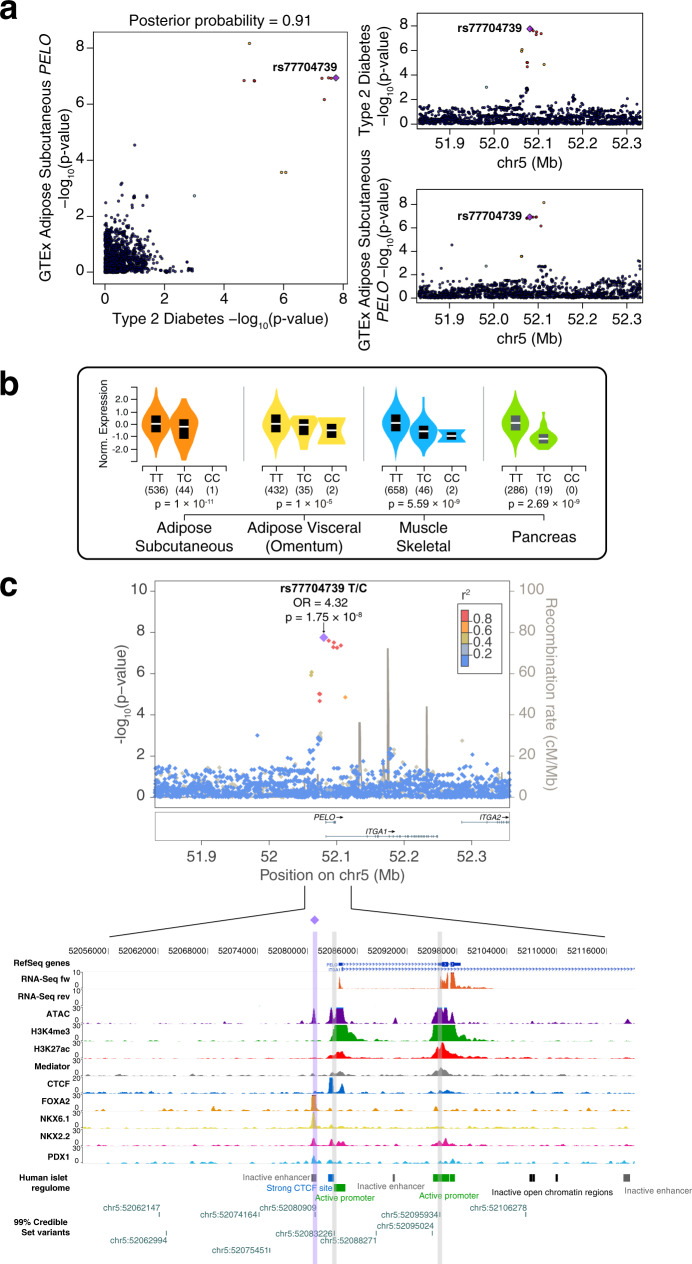
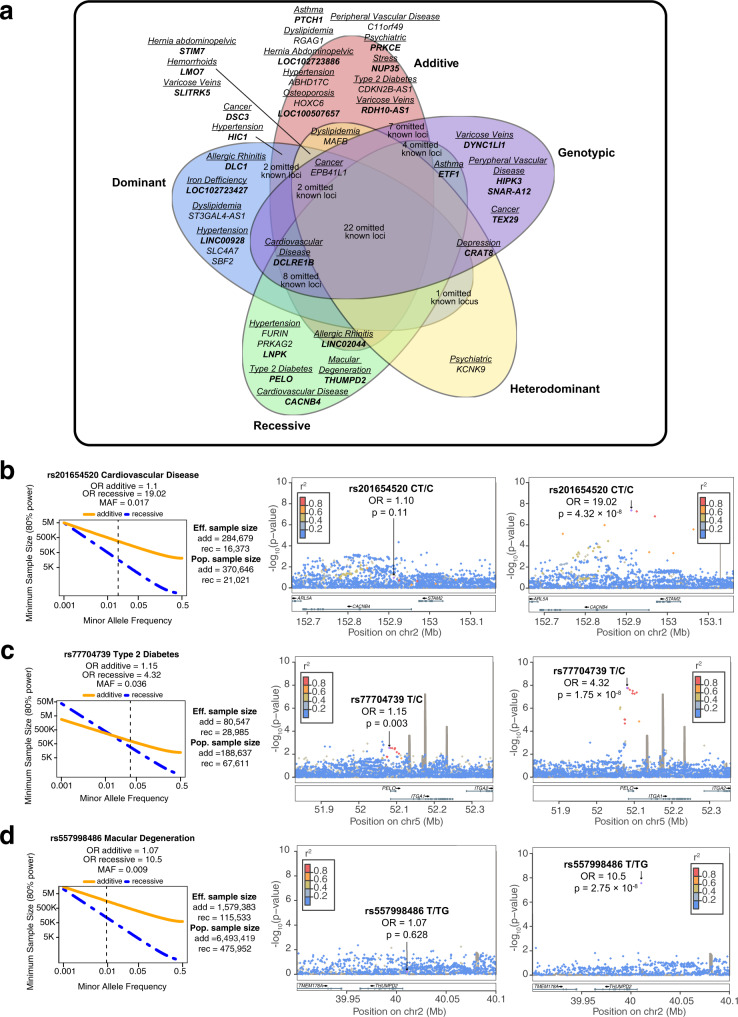
Genome-wide association studies (GWAS) are not fully comprehensive, as current strategies typically test only the additive model, exclude the X chromosome, and use only one reference panel for genotype imputation. We implement an extensive GWAS strategy, GUIDANCE, which improves genotype imputation by using multiple reference panels and includes the analysis of the X chromosome and non-additive models to test for association. We apply this methodology to 62,281 subjects across 22 age-related diseases and identify 94 genome-wide associated loci, including 26 previously unreported. Moreover, we observe that 27.7% of the 94 loci are missed if we use standard imputation strategies with a single reference panel, such as HRC, and only test the additive model. Among the new findings, we identify three novel low-frequency recessive variants with odds ratios larger than 4, which need at least a three-fold larger sample size to be detected under the additive model. This study highlights the benefits of applying innovative strategies to better uncover the genetic architecture of complex diseases.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
