

Generation of a humanized Aβ expressing mouse demonstrating aspects of Alzheimer's disease-like pathology
- PMID: 33893290
- PMCID: PMC8065162
- DOI: 10.1038/s41467-021-22624-z
Generation of a humanized Aβ expressing mouse demonstrating aspects of Alzheimer's disease-like pathology
Abstract
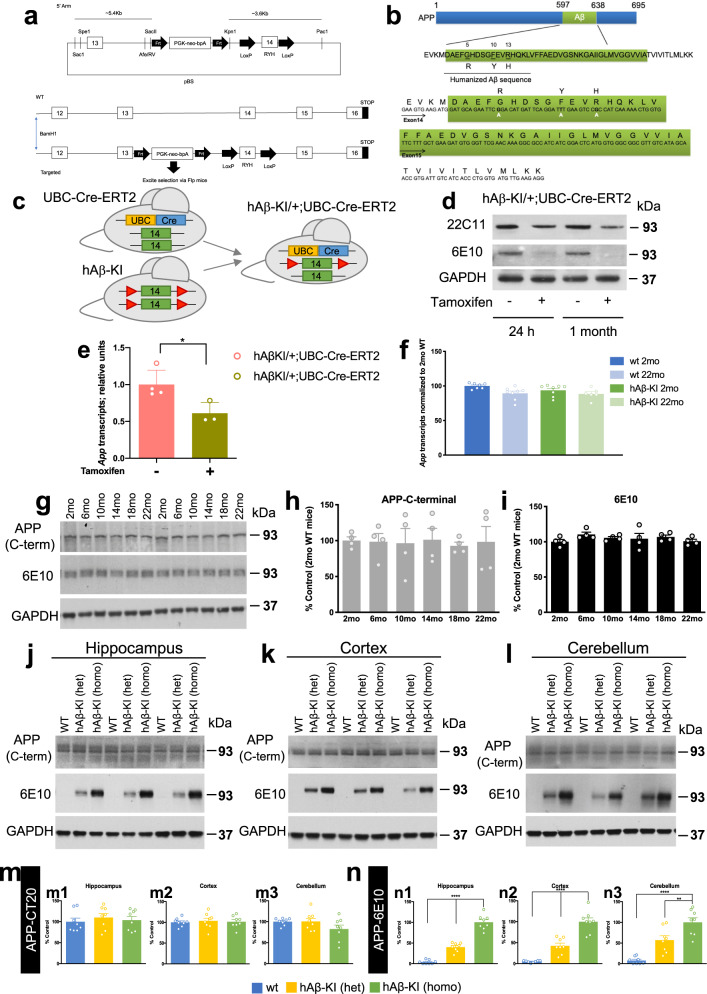
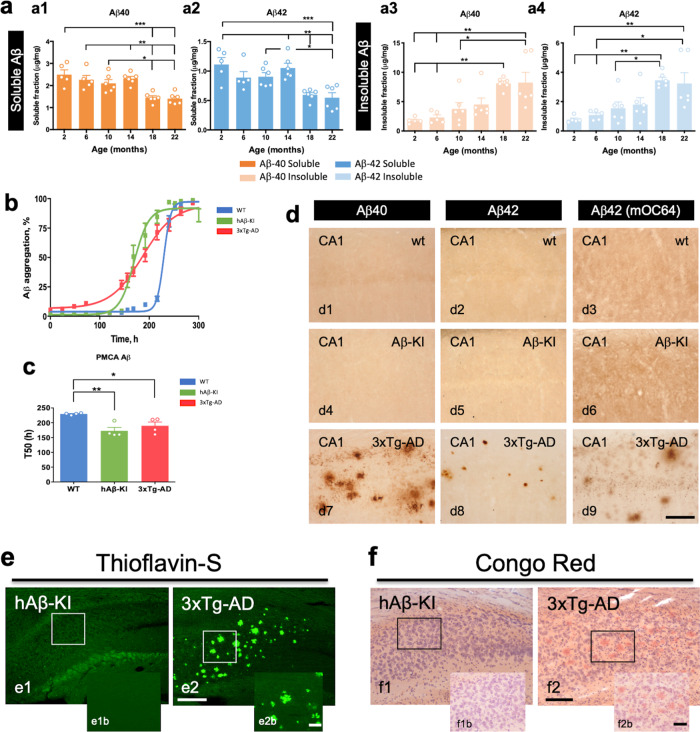
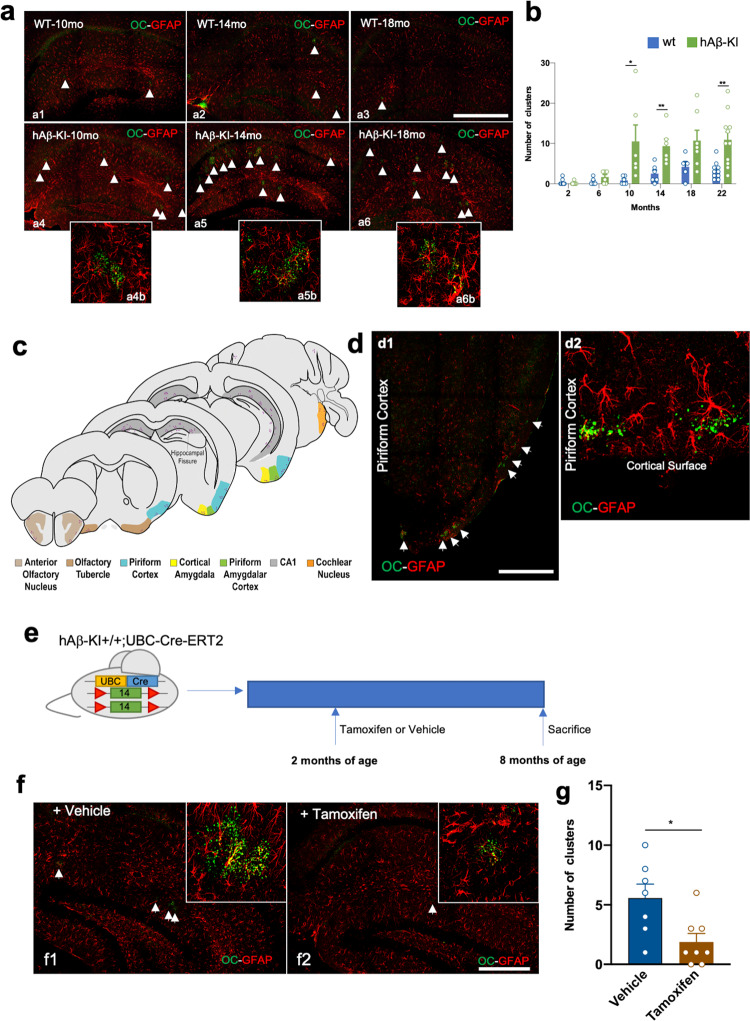
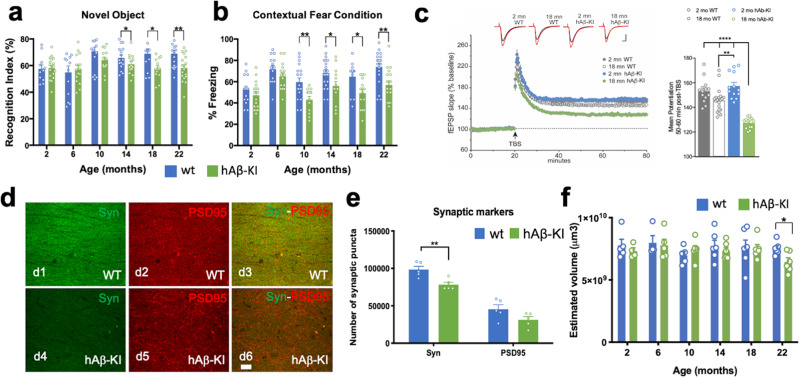
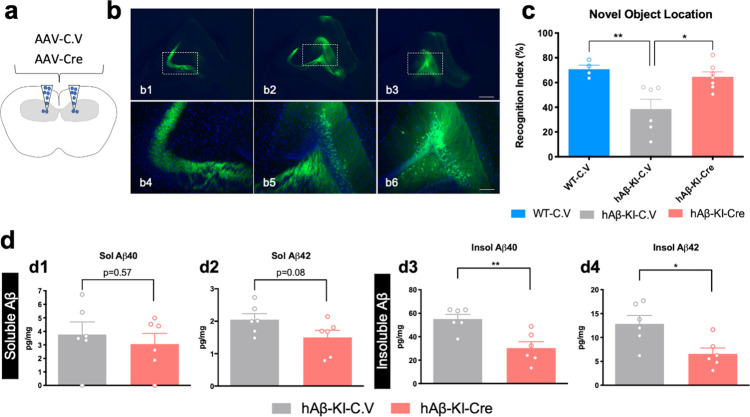
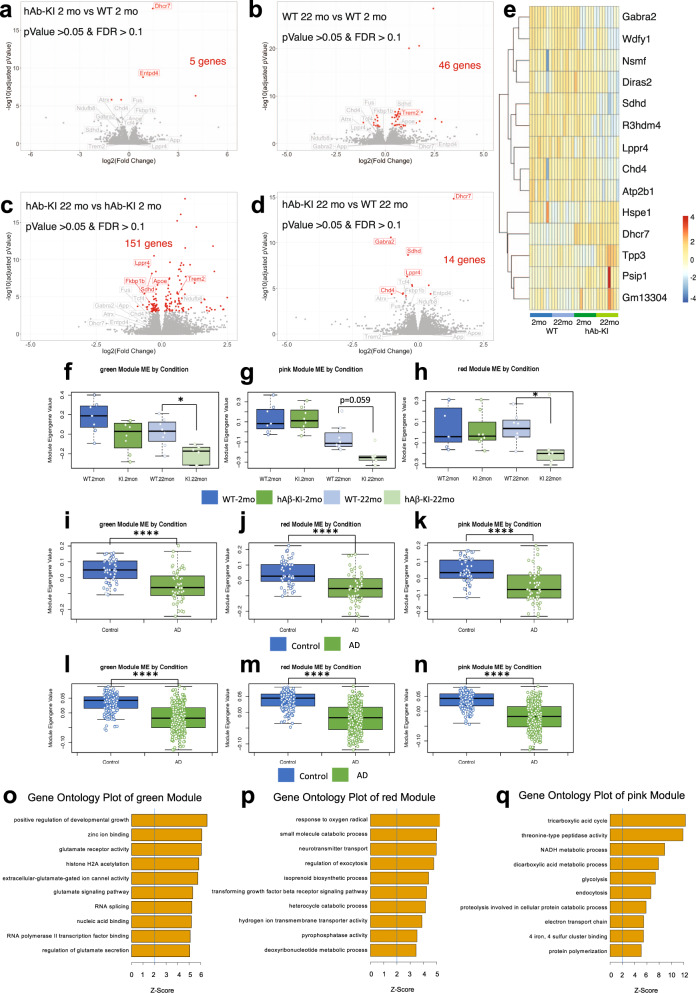
The majority of Alzheimer's disease (AD) cases are late-onset and occur sporadically, however most mouse models of the disease harbor pathogenic mutations, rendering them better representations of familial autosomal-dominant forms of the disease. Here, we generated knock-in mice that express wildtype human Aβ under control of the mouse App locus. Remarkably, changing 3 amino acids in the mouse Aβ sequence to its wild-type human counterpart leads to age-dependent impairments in cognition and synaptic plasticity, brain volumetric changes, inflammatory alterations, the appearance of Periodic Acid-Schiff (PAS) granules and changes in gene expression. In addition, when exon 14 encoding the Aβ sequence was flanked by loxP sites we show that Cre-mediated excision of exon 14 ablates hAβ expression, rescues cognition and reduces the formation of PAS granules.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
