

Health impact of tafamidis in transthyretin amyloid cardiomyopathy patients: an analysis from the Tafamidis in Transthyretin Cardiomyopathy Clinical Trial (ATTR-ACT) and the open-label long-term extension studies
- PMID: 33895806
- PMCID: PMC9382662
- DOI: 10.1093/ehjqcco/qcab031
Health impact of tafamidis in transthyretin amyloid cardiomyopathy patients: an analysis from the Tafamidis in Transthyretin Cardiomyopathy Clinical Trial (ATTR-ACT) and the open-label long-term extension studies
Abstract
Aim: The Tafamidis in Transthyretin Cardiomyopathy Clinical Trial (ATTR-ACT) showed that tafamidis reduced all-cause mortality and cardiovascular-related hospitalizations in patients with transthyretin amyloid cardiomyopathy (ATTR-CM). This study aimed to estimate the impact of tafamidis on survival and quality-adjusted life-years (QALYs).
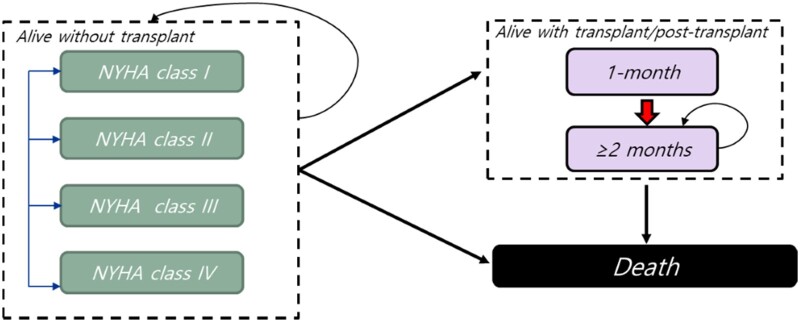
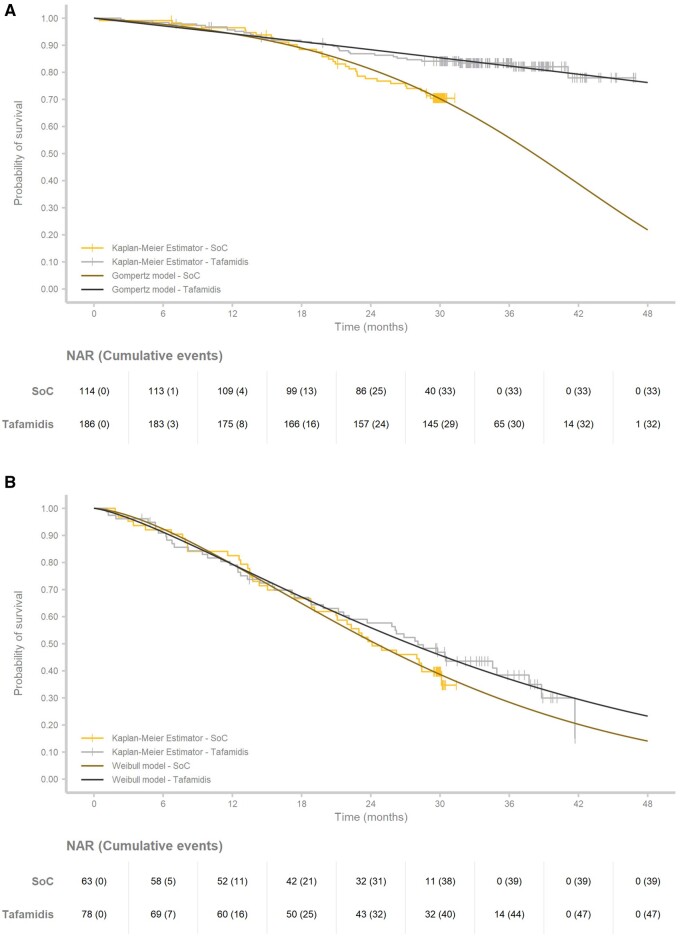
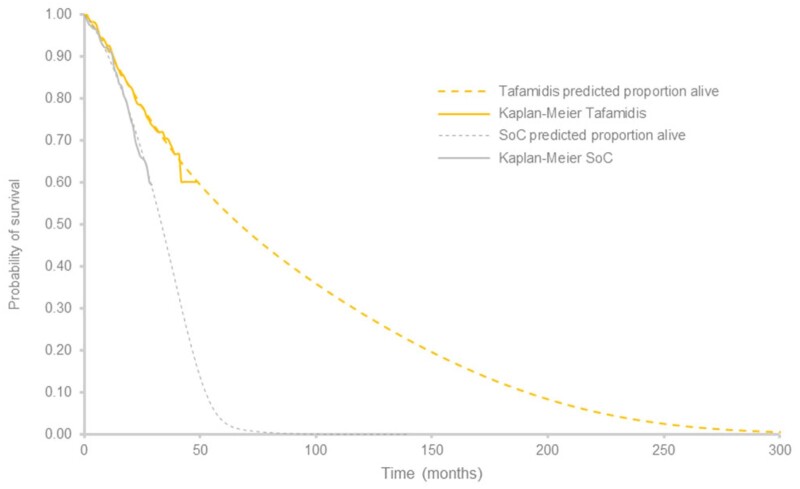
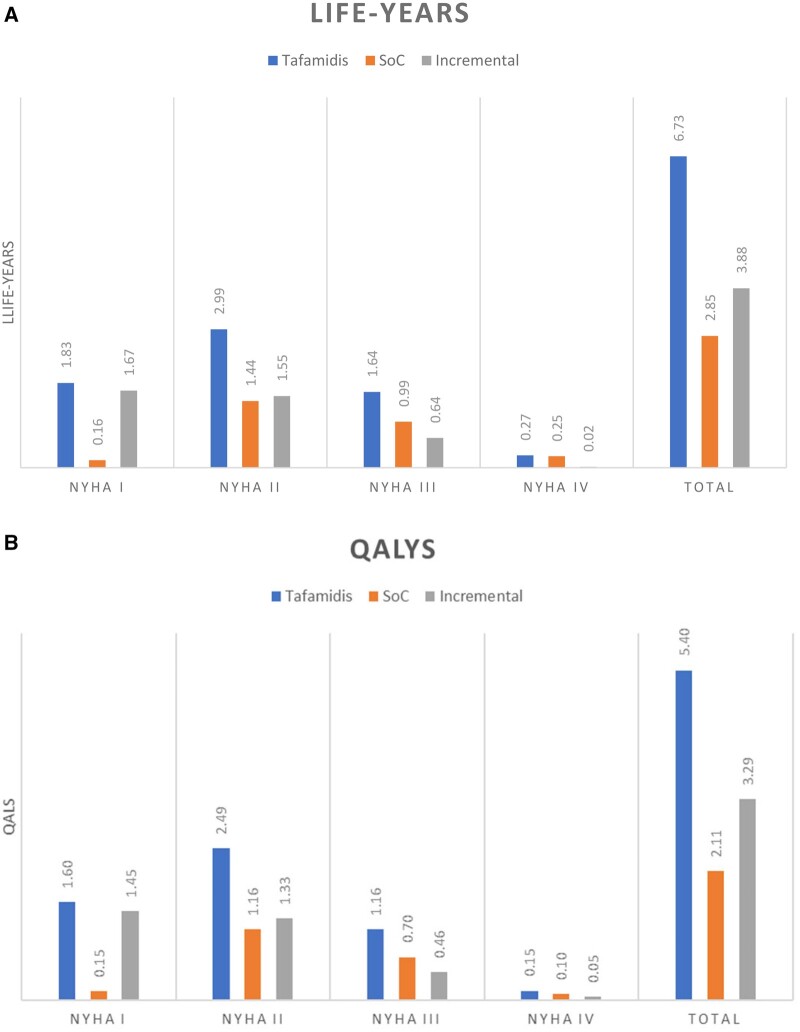
Methods and results: A multi-state, cohort, Markov model was developed to simulate the disease course of ATTR-CM throughout a lifetime. For survival extrapolation, survival curves were fitted by treatment arm and New York Heart Association (NYHA) Class I/II (68% of patients) and NYHA Class III (32% of patients) cohorts using the individual patient-level data from both the ATTR-ACT and the corresponding long-term extension study. Univariate and multivariate sensitivity analyses were conducted. The predicted mean survival for the total population (NYHA Class I/II + III) was 6.73 years for tafamidis and 2.85 years for the standard of care (SoC), resulting in an incremental mean survival of 3.88 years [95% confidence interval (CI) 1.32-5.66]. Of the 6.73 life-years, patients on tafamidis spend, on average, 4.82 years in NYHA Class I/II, while patients on SoC spend an average of 1.60 life-years in these classes. The combination of longer survival in lower NYHA classes produced a QALY gain of 5.39 for tafamidis and 2.11 for SoC, resulting in 3.29 incremental QALYs (95% CI 1.21-4.74) in favour of tafamidis.
Conclusion: Based on the disease simulation model results, tafamidis is expected to more than double the life expectancy and QALYs of ATTR-CM patients compared to SoC. Longer-term follow-up data from the ATTR-ACT extension study will further inform these findings.
Clinical trials.gov identifier: NCT01994889 (date of registration: 26 November 2013).
Keywords: Cardiomyopathy; Mortality; Tafamidis; Transthyretin; Amyloidosis.
© The Author(s) 2021. Published by Oxford University Press on behalf of the European Society of Cardiology.
Figures
References
-
- Rapezzi C, Quarta CC, Riva L, Longhi S, Gallelli I, Lorenzini M. et al. Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol 2010;7:398–408. - PubMed
-
- Arbustini E, Merlini G.. Early identification of transthyretin-related hereditary cardiac amyloidosis. JACC Cardiovasc Imaging 2014;7:511–514. - PubMed
-
- Barrett CD, Alexander KM, Zhao H, Haddad F, Cheng P, Liao R. et al. Outcomes in patients with cardiac amyloidosis undergoing heart transplantation. JACC Heart Fail 2020;8:461–468. - PubMed
-
- Ericzon B-G, Wilczek HE, Larsson M, Wijayatunga P, Stangou A, Pena JR. et al. Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation 2015;99:1847–1854. - PubMed
-
- Ruberg FL, Maurer MS, Judge DP, Zeldenrust S, Skinner M, Kim AY. et al. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the Transthyretin Amyloidosis Cardiac Study (TRACS). American Heart Journal 2012;164:222–228. - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
