

Genomic insights into the conservation status of the world's last remaining Sumatran rhinoceros populations
- PMID: 33896938
- PMCID: PMC8071806
- DOI: 10.1038/s41467-021-22386-8
Genomic insights into the conservation status of the world's last remaining Sumatran rhinoceros populations
Abstract
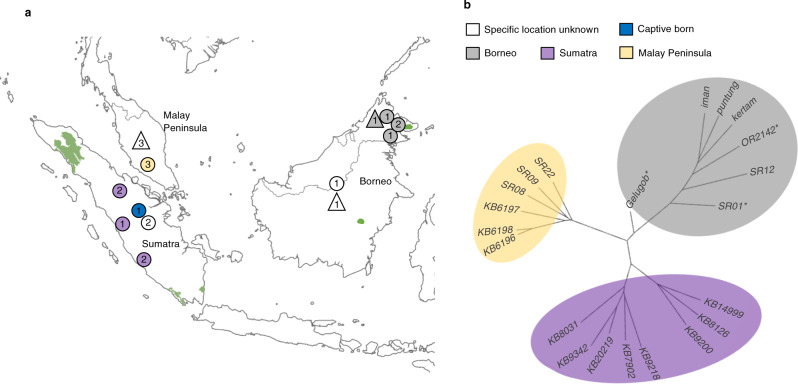
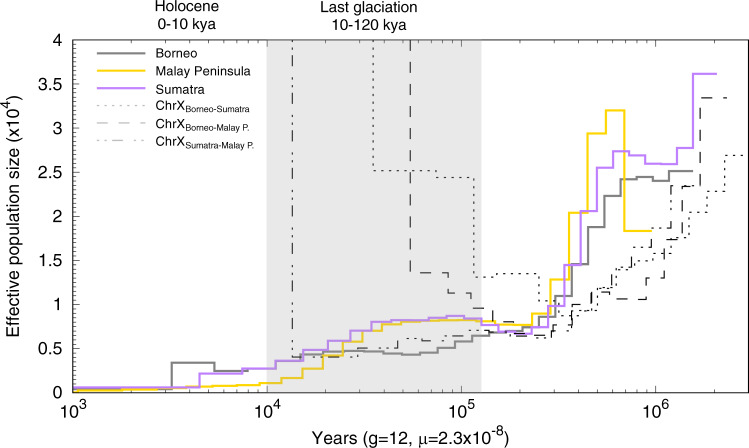
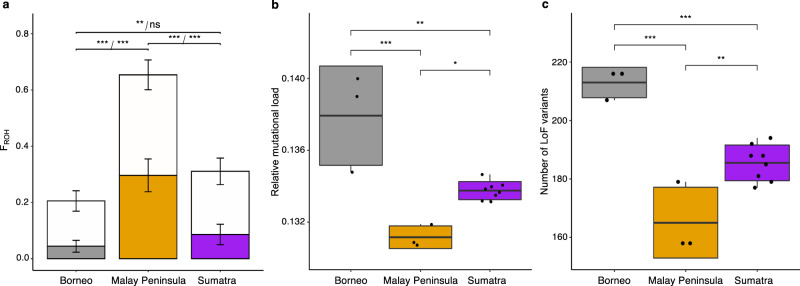
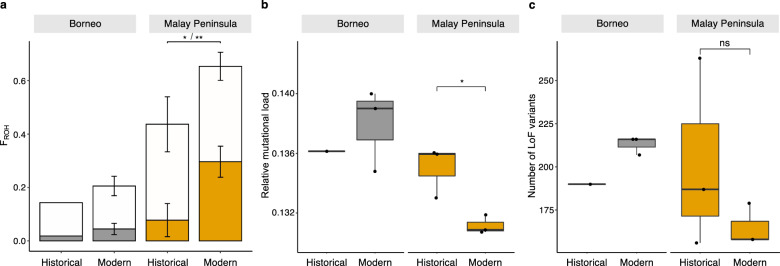
Small populations are often exposed to high inbreeding and mutational load that can increase the risk of extinction. The Sumatran rhinoceros was widespread in Southeast Asia, but is now restricted to small and isolated populations on Sumatra and Borneo, and most likely extinct on the Malay Peninsula. Here, we analyse 5 historical and 16 modern genomes from these populations to investigate the genomic consequences of the recent decline, such as increased inbreeding and mutational load. We find that the Malay Peninsula population experienced increased inbreeding shortly before extirpation, which possibly was accompanied by purging. The populations on Sumatra and Borneo instead show low inbreeding, but high mutational load. The currently small population sizes may thus in the near future lead to inbreeding depression. Moreover, we find little evidence for differences in local adaptation among populations, suggesting that future inbreeding depression could potentially be mitigated by assisted gene flow among populations.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
