

Immune Subtyping in Latent Tuberculosis
- PMID: 33897680
- PMCID: PMC8059438
- DOI: 10.3389/fimmu.2021.595746
Immune Subtyping in Latent Tuberculosis
Abstract
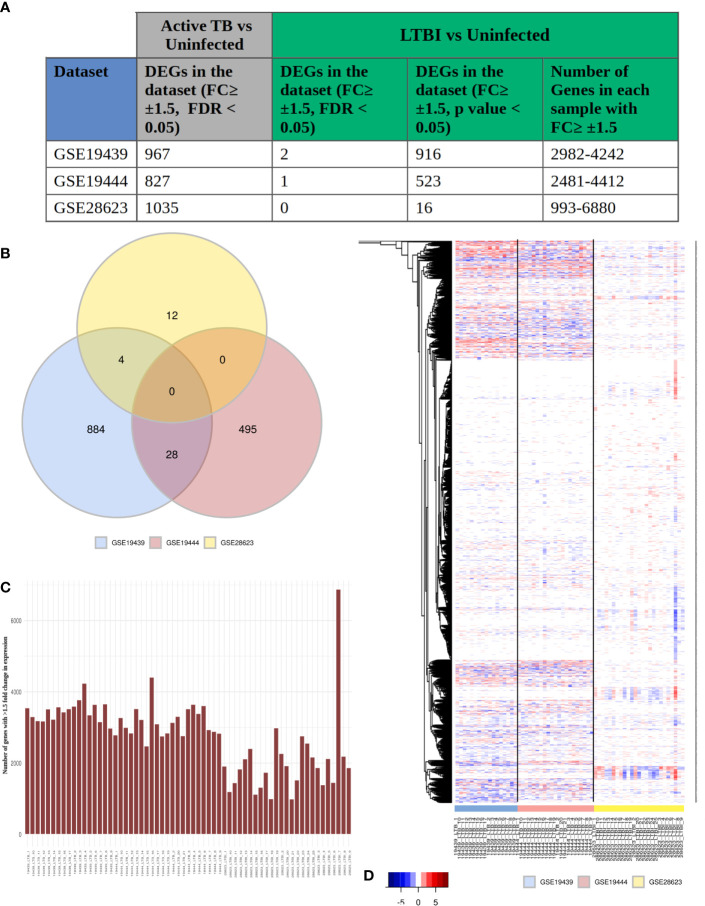
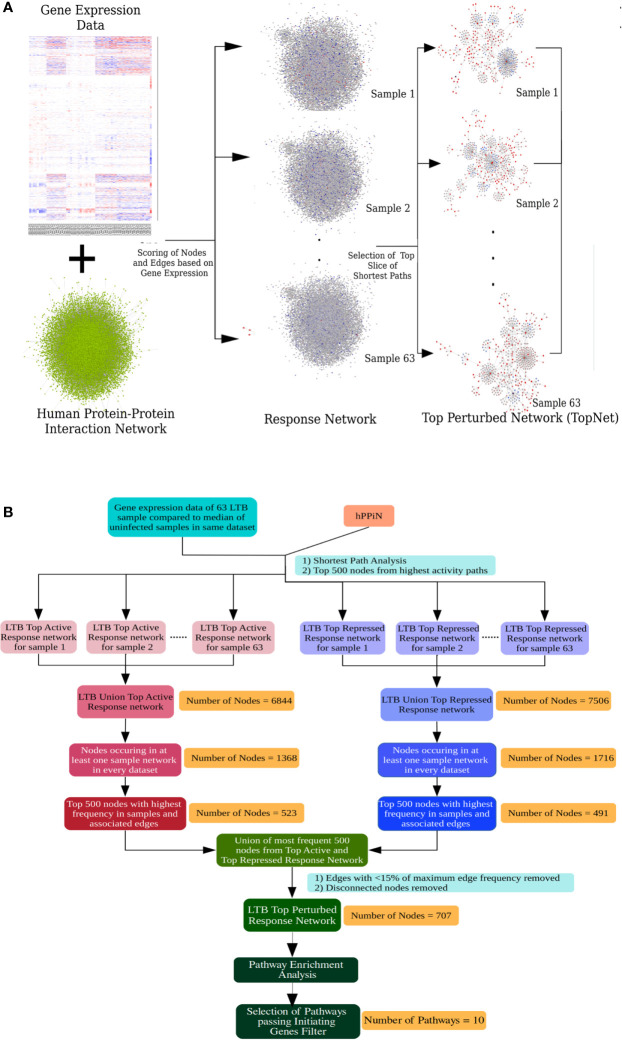
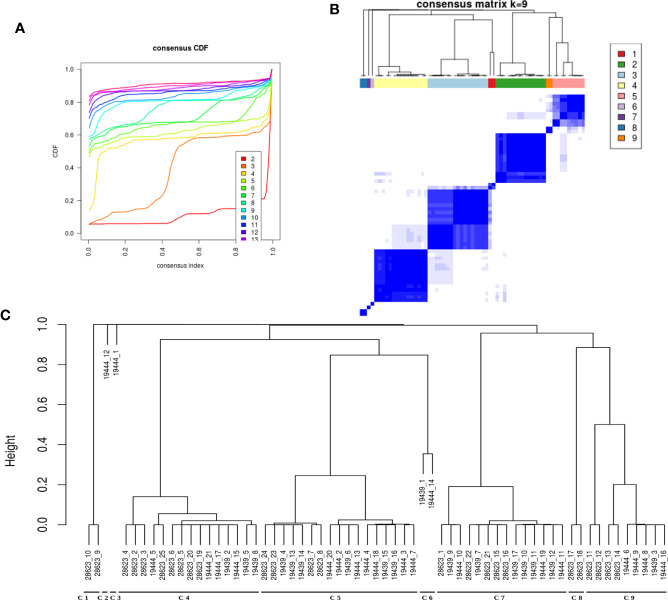
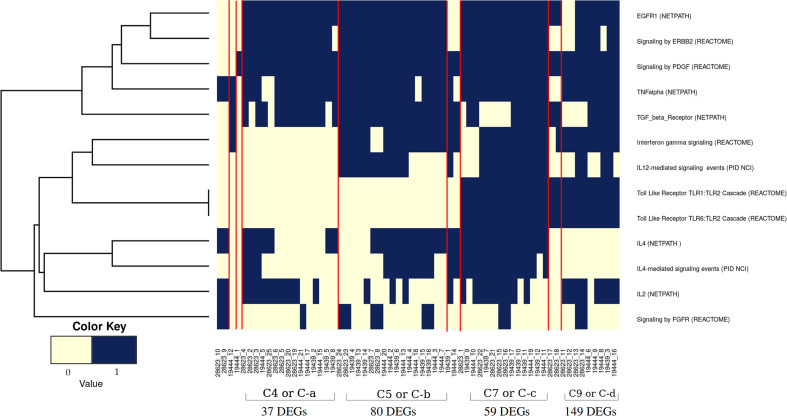
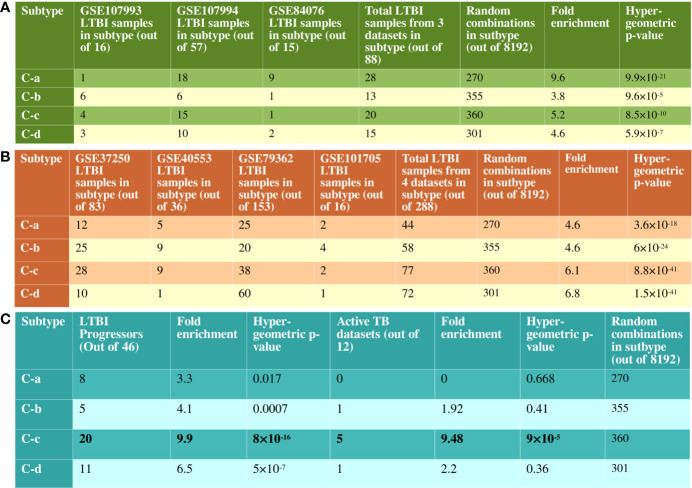
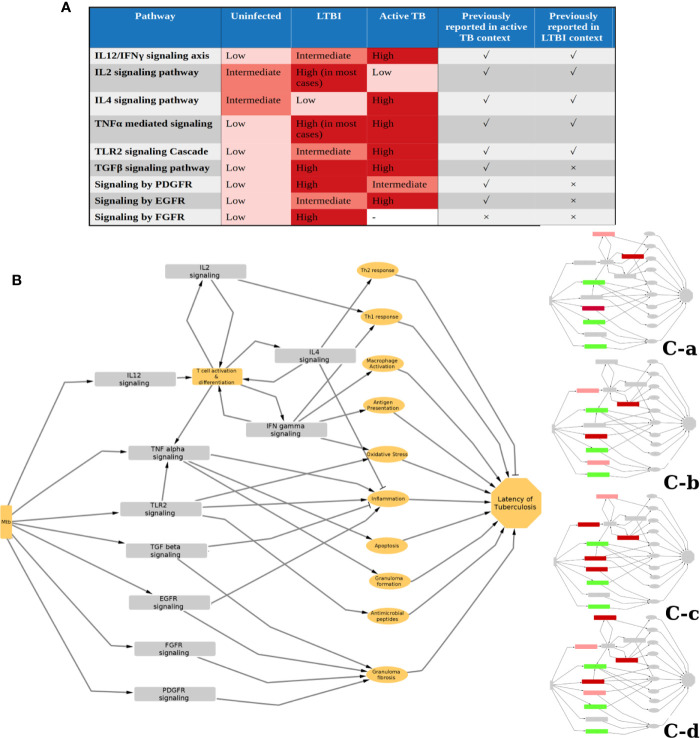
Latent tuberculosis infection (LTBI) poses a major roadblock in the global effort to eradicate tuberculosis (TB). A deep understanding of the host responses involved in establishment and maintenance of TB latency is required to propel the development of sensitive methods to detect and treat LTBI. Given that LTBI individuals are typically asymptomatic, it is challenging to differentiate latently infected from uninfected individuals. A major contributor to this problem is that no clear pattern of host response is linked with LTBI, as molecular correlates of latent infection have been hard to identify. In this study, we have analyzed the global perturbations in host response in LTBI individuals as compared to uninfected individuals and particularly the heterogeneity in such response, across LTBI cohorts. For this, we constructed individualized genome-wide host response networks informed by blood transcriptomes for 136 LTBI cases and have used a sensitive network mining algorithm to identify top-ranked host response subnetworks in each case. Our analysis indicates that despite the high heterogeneity in the gene expression profiles among LTBI samples, clear patterns of perturbation are found in the immune response pathways, leading to grouping LTBI samples into 4 different immune-subtypes. Our results suggest that different subnetworks of molecular perturbations are associated with latent tuberculosis.
Keywords: genome-wide network analysis; heterogeneity; immune subtypes; latent tuberculosis; transcriptomics.
Copyright © 2021 Banerjee, Baloni, Singh and Chandra.
Conflict of interest statement
NC is a co-founder of the companies qBiome Research Pvt Ltd and HealthSeq Precision Medicine Pvt Ltd. They had no role in this manuscript. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- WHO . WHO | Global tuberculosis report 2018. (2018).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical