

Enzyme dynamics: Looking beyond a single structure
- PMID: 33897908
- PMCID: PMC8064270
- DOI: 10.1002/cctc.202000665
Enzyme dynamics: Looking beyond a single structure
Abstract
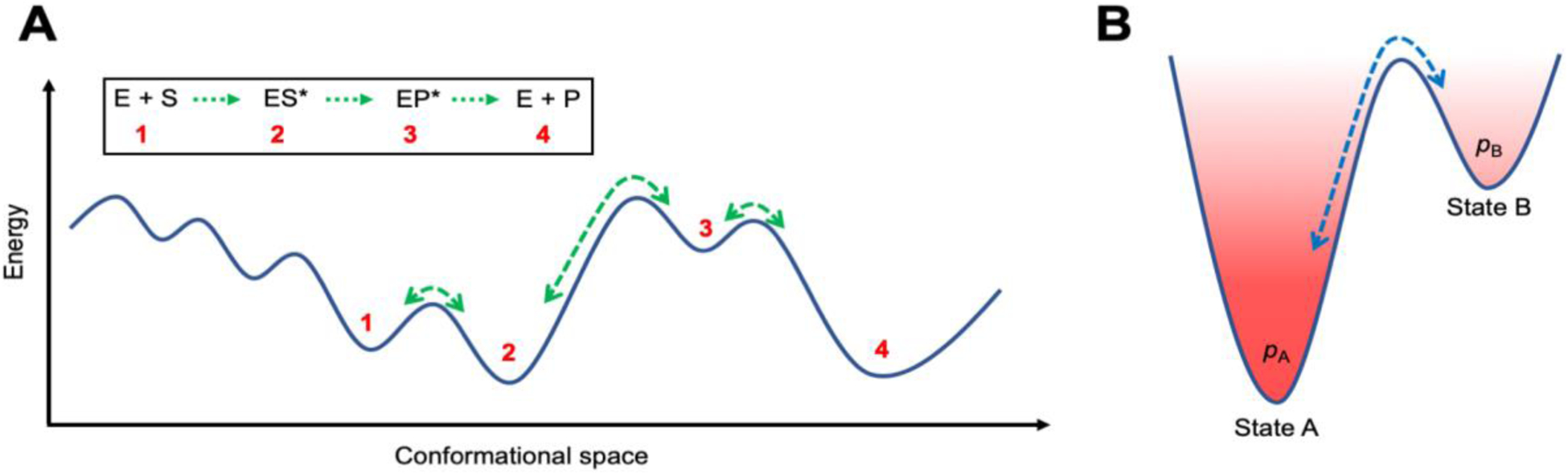
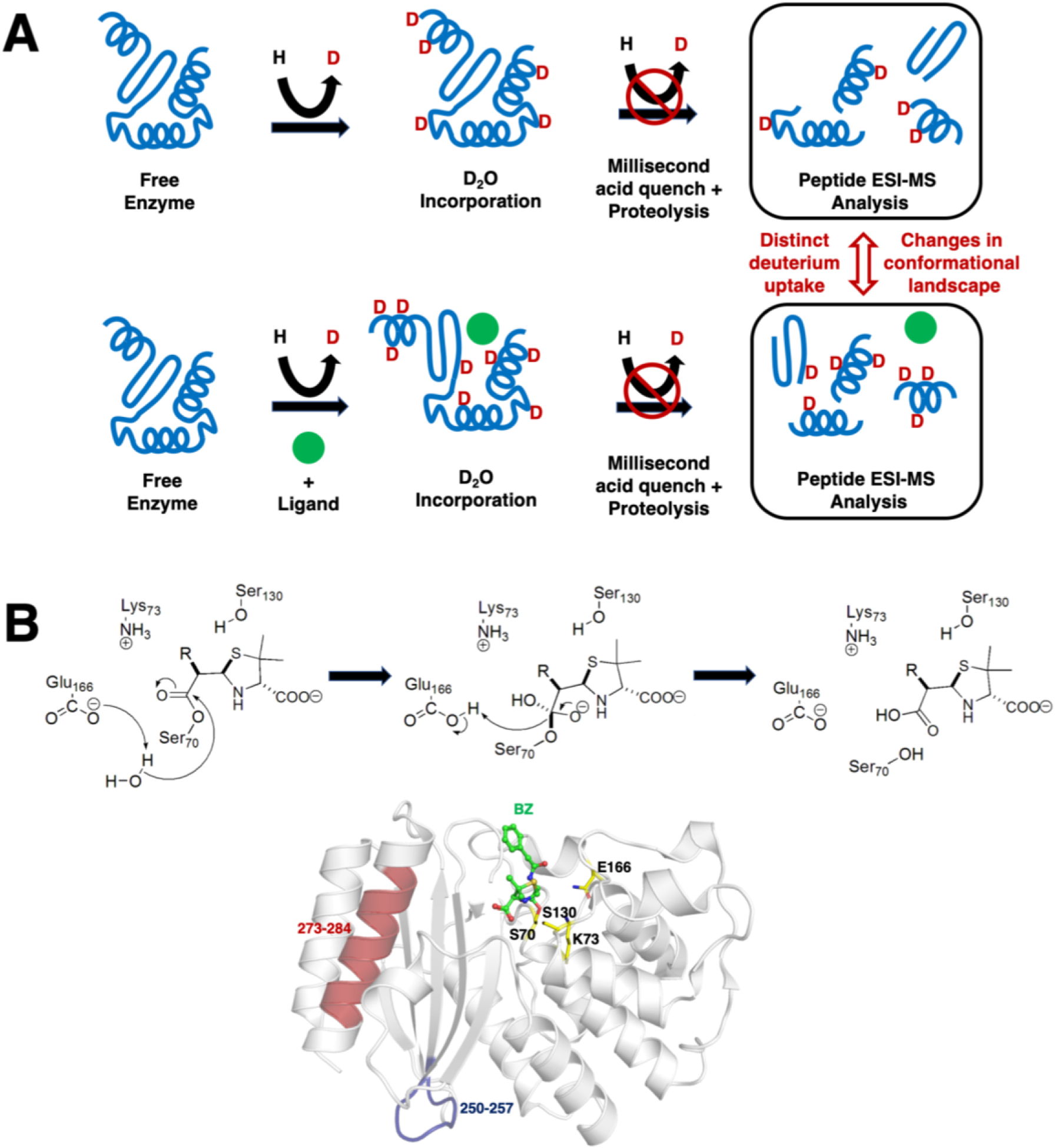
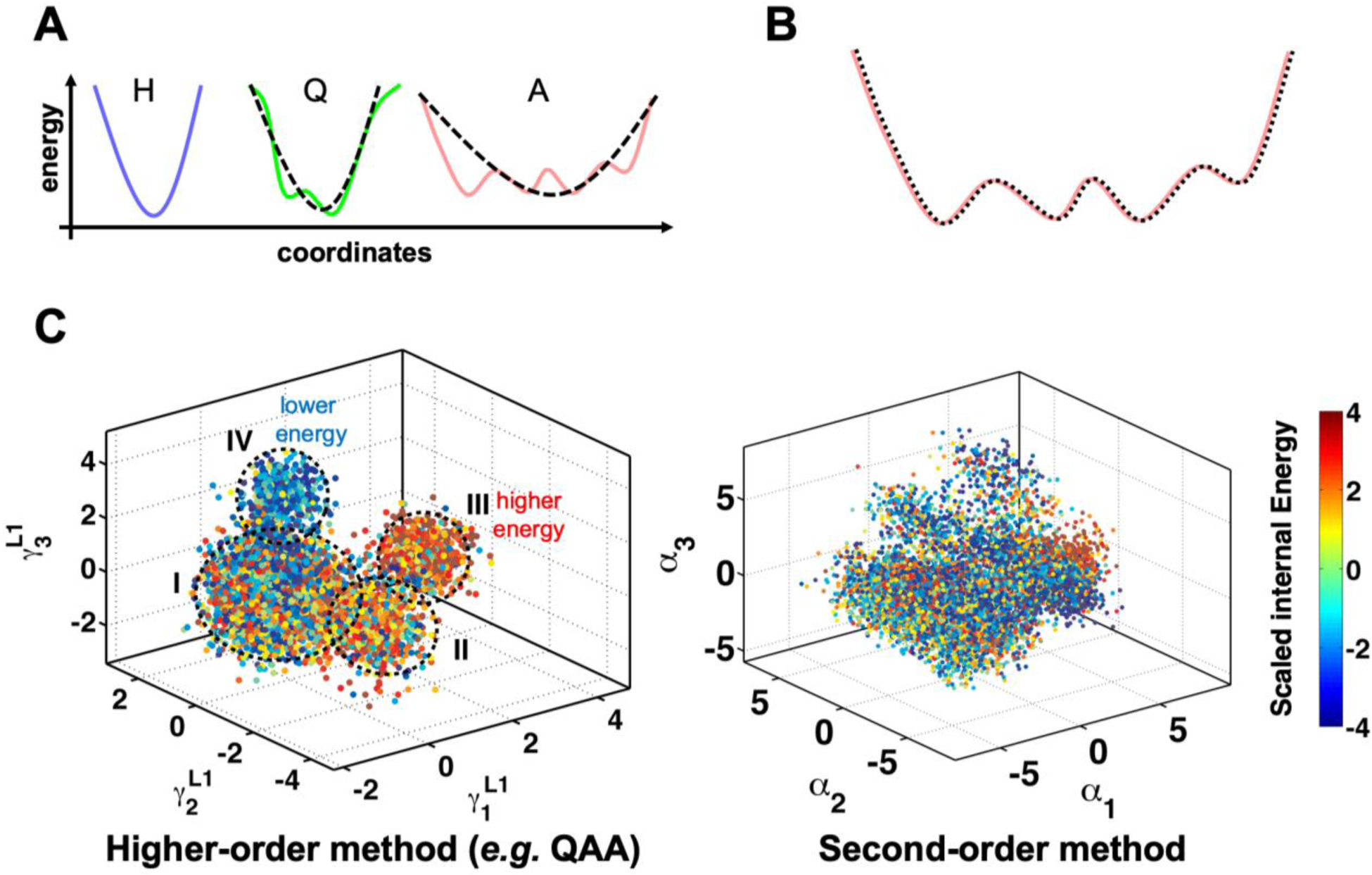
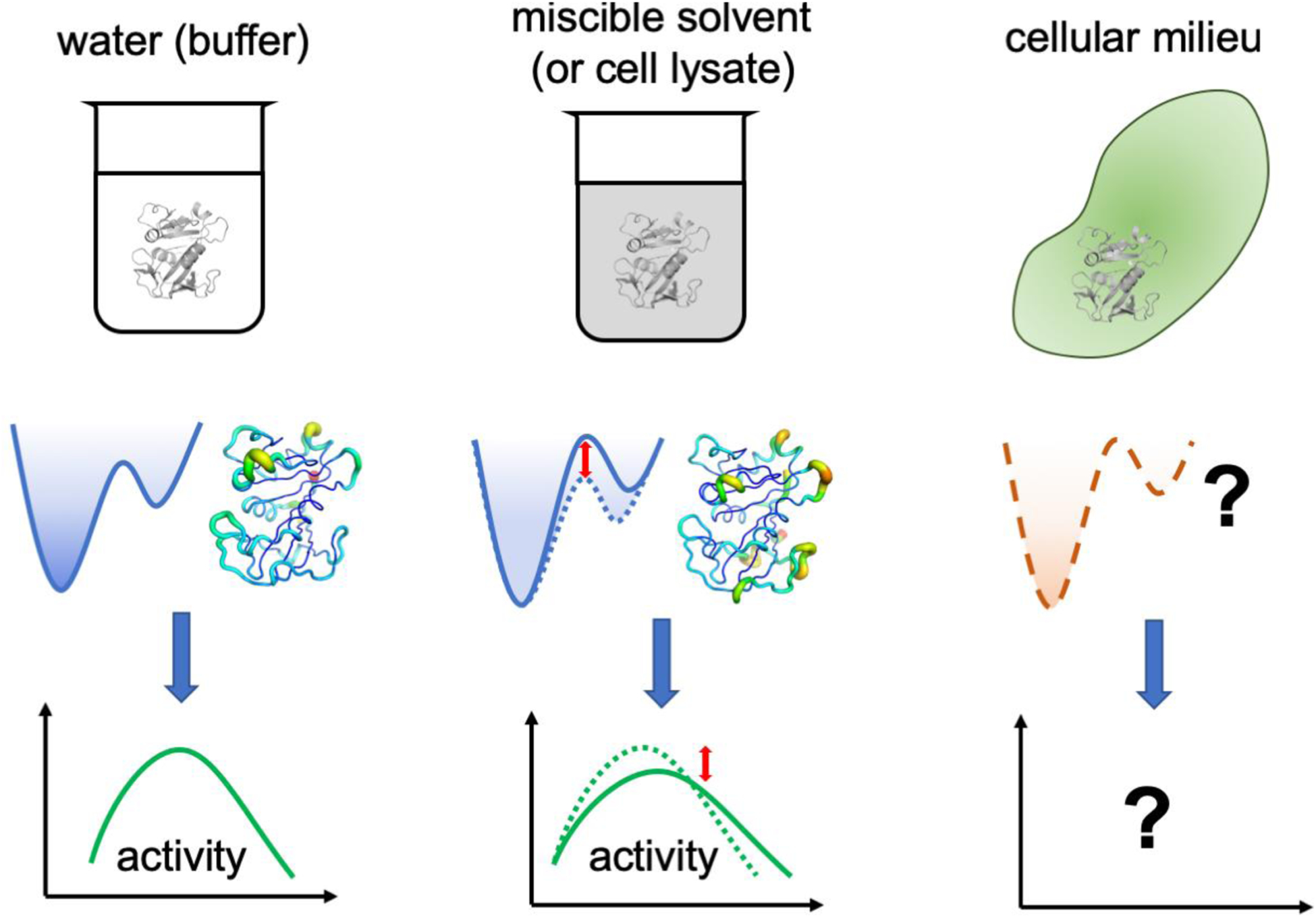
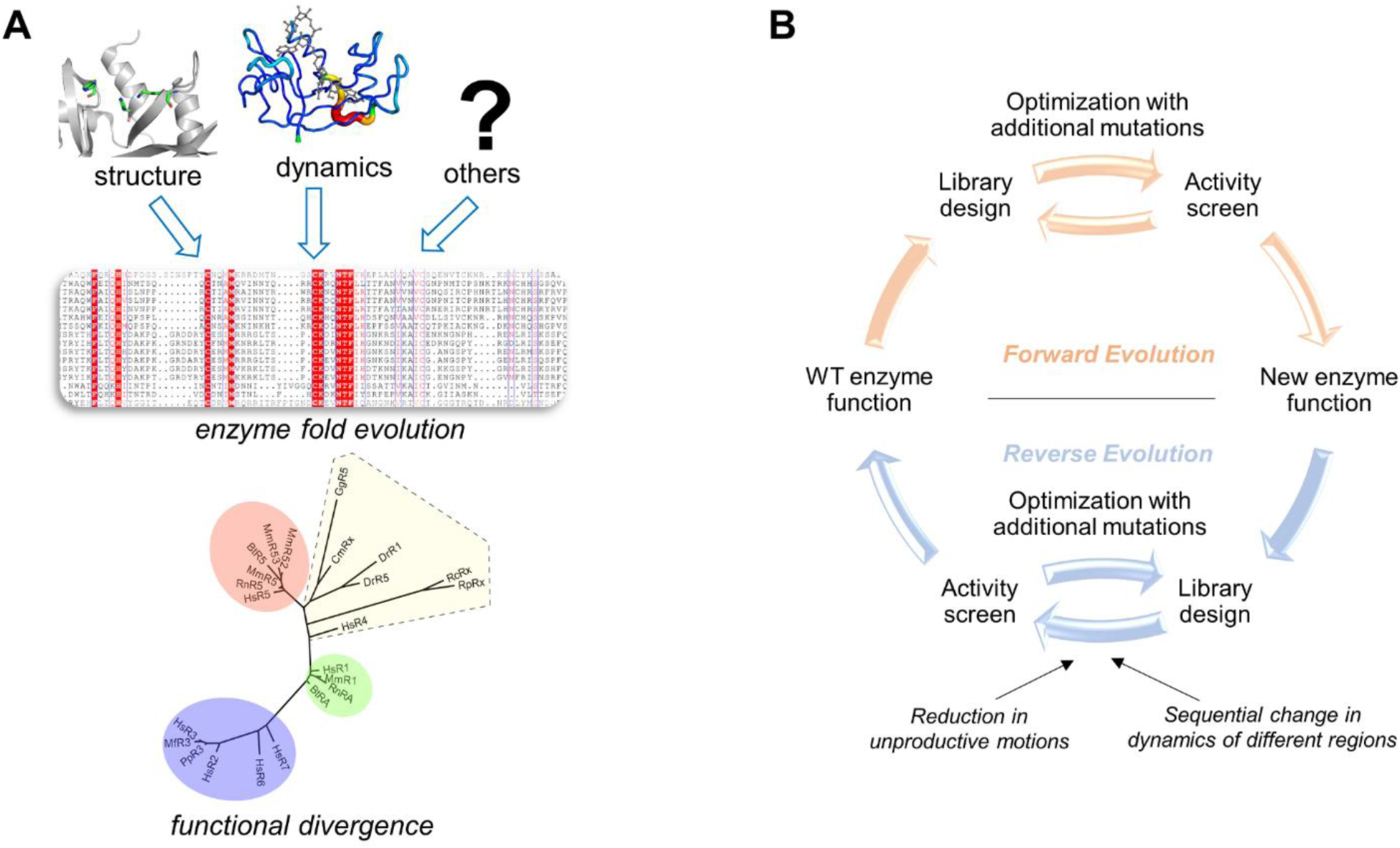
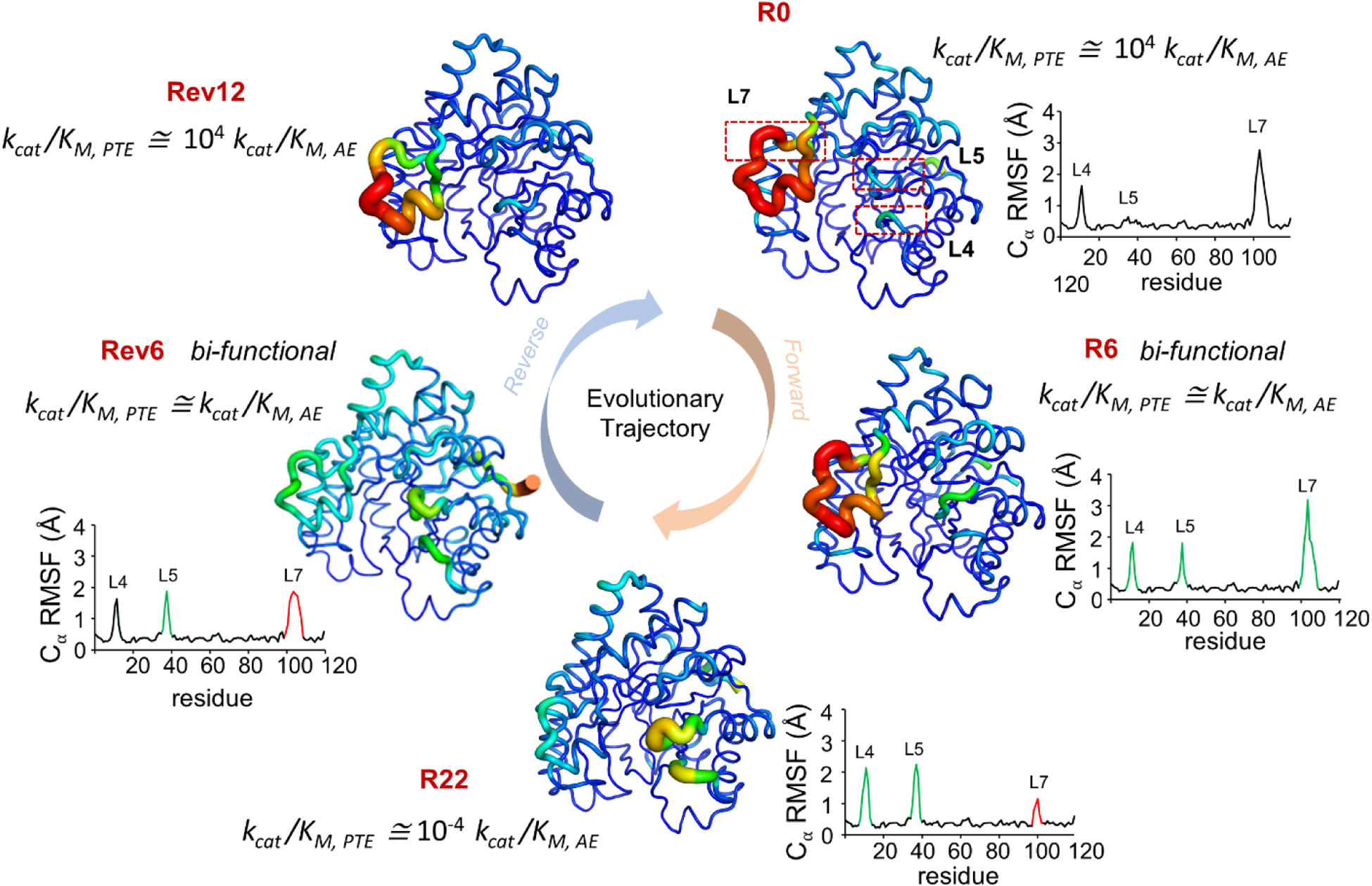
Conventional understanding of how enzymes function strongly emphasizes the role of structure. However, increasing evidence clearly indicates that enzymes do not remain fixed or operate exclusively in or close to their native structure. Different parts of the enzyme (from individual residues to full domains) undergo concerted motions on a wide range of time-scales, including that of the catalyzed reaction. Information obtained on these internal motions and conformational fluctuations has so far uncovered and explained many aspects of enzyme mechanisms, which could not have been understood from a single structure alone. Although there is wide interest in understanding enzyme dynamics and its role in catalysis, several challenges remain. In addition to technical difficulties, the vast majority of investigations are performed in dilute aqueous solutions, where conditions are significantly different than the cellular milieu where a large number of enzymes operate. In this review, we discuss recent developments, several challenges as well as opportunities related to this topic. The benefits of considering dynamics as an integral part of the enzyme function can also enable new means of biocatalysis, engineering enzymes for industrial and medicinal applications.
Keywords: Biocatalysis; conformational sub-states; directed evolution; enzyme engineering; protein dynamics.
Figures
Similar articles
-
A Biophysical Perspective on Enzyme Catalysis.Biochemistry. 2019 Feb 12;58(6):438-449. doi: 10.1021/acs.biochem.8b01004. Epub 2018 Dec 18. Biochemistry. 2019. PMID: 30507164 Free PMC article.
-
Protein conformational populations and functionally relevant substates.Acc Chem Res. 2014 Jan 21;47(1):149-56. doi: 10.1021/ar400084s. Epub 2013 Aug 29. Acc Chem Res. 2014. PMID: 23988159 Review.
-
Enzymes: An integrated view of structure, dynamics and function.Microb Cell Fact. 2006 Jan 12;5:2. doi: 10.1186/1475-2859-5-2. Microb Cell Fact. 2006. PMID: 16409630 Free PMC article.
-
Role of protein dynamics in reaction rate enhancement by enzymes.J Am Chem Soc. 2005 Nov 2;127(43):15248-56. doi: 10.1021/ja055251s. J Am Chem Soc. 2005. PMID: 16248667
-
Role of dynamics in enzyme catalysis: substantial versus semantic controversies.Acc Chem Res. 2015 Feb 17;48(2):466-73. doi: 10.1021/ar500322s. Epub 2014 Dec 24. Acc Chem Res. 2015. PMID: 25539442 Free PMC article. Review.
Cited by
-
Editing Domain Motions Preorganize the Synthetic Active Site of Prolyl-tRNA Synthetase.ACS Catal. 2020 Sep 4;10(17):10229-10242. doi: 10.1021/acscatal.0c02381. Epub 2020 Aug 14. ACS Catal. 2020. PMID: 34295570 Free PMC article.
-
From random to rational: improving enzyme design through electric fields, second coordination sphere interactions, and conformational dynamics.Chem Sci. 2023 Sep 13;14(40):10997-11011. doi: 10.1039/d3sc02982d. eCollection 2023 Oct 18. Chem Sci. 2023. PMID: 37860658 Free PMC article. Review.
-
Enzymes in a human cytoplasm model organize into submetabolon complexes.Proc Natl Acad Sci U S A. 2025 Feb 4;122(5):e2414206122. doi: 10.1073/pnas.2414206122. Epub 2025 Jan 28. Proc Natl Acad Sci U S A. 2025. PMID: 39874290 Free PMC article.
-
Modeling Ensembles of Enzyme Reaction Pathways with Hi-MSM Reveals the Importance of Accounting for Pathway Diversity.J Phys Chem B. 2022 Dec 1;126(47):9748-9758. doi: 10.1021/acs.jpcb.2c04496. Epub 2022 Nov 16. J Phys Chem B. 2022. PMID: 36383711 Free PMC article.
-
Conformational Modulation of a Mobile Loop Controls Catalysis in the (βα)8-Barrel Enzyme of Histidine Biosynthesis HisF.JACS Au. 2024 Aug 15;4(8):3258-3276. doi: 10.1021/jacsau.4c00558. eCollection 2024 Aug 26. JACS Au. 2024. PMID: 39211614 Free PMC article.
References
-
- Boehr DD, McElheny D, Dyson HJ, Wright PE, Science 2006, 313, 1638–1642. - PubMed
-
- Benkovic SJ, Hammes GG, Hammes-Schiffer S, Biochemistry 2008, 47, 3317–3321. - PubMed
-
- Ramanathan A, Savol A, Burger V, Chennubhotla C, Agarwal PK, Acc. Chem. Res 2014, 47, 149–156. - PubMed
-
- Dahiyat BI, Mayo SL, Science 1997, 278, 82–87. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources