

tSFM 1.0: tRNA Structure-Function Mapper
- PMID: 33904572
- PMCID: PMC8545343
- DOI: 10.1093/bioinformatics/btab247
tSFM 1.0: tRNA Structure-Function Mapper
Abstract
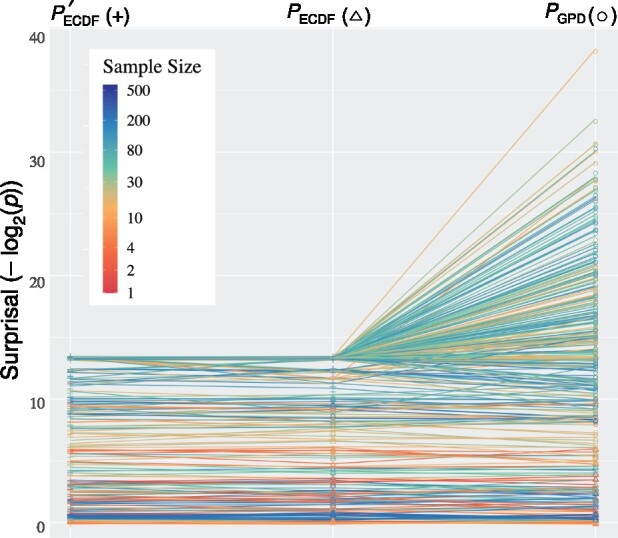
Motivation: Structure-conditioned information statistics have proven useful to predict and visualize tRNA Class-Informative Features (CIFs) and their evolutionary divergences. Although permutation P-values can quantify the significance of CIF divergences between two taxa, their naive Monte Carlo approximation is slow and inaccurate. The Peaks-over-Threshold approach of Knijnenburg et al. (2009) promises improvements to both speed and accuracy of permutation P-values, but has no publicly available API.
Results: We present tRNA Structure-Function Mapper (tSFM) v1.0, an open-source, multi-threaded application that efficiently computes, visualizes and assesses significance of single- and paired-site CIFs and their evolutionary divergences for any RNA, protein, gene or genomic element sequence family. Multiple estimators of permutation P-values for CIF evolutionary divergences are provided along with confidence intervals. tSFM is implemented in Python 3 with compiled C extensions and is freely available through GitHub (https://github.com/tlawrence3/tSFM) and PyPI.
Availability and implementation: The data underlying this article are available on GitHub at https://github.com/tlawrence3/tSFM.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2021. Published by Oxford University Press. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
References
-
- Benjamini Y., Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat Soc. Ser. B, 57, 289–300.
-
- Campbell B. et al. (2016) Application of the envelope peaks over threshold (EPOT) method for probabilistic assessment of dynamic stability. Ocean Eng., 120, 298–304.
-
- Collins-Hed A.I., Ardell D.H. (2019) Match fitness landscapes for macromolecular interaction networks: selection for translational accuracy and rate can displace tRNA-binding interfaces of non-cognate aminoacyl-tRNA synthetases. Theor. Popul. Biol., 129, 68–80. - PubMed
-
- Freyhult E. et al. (2007) New computational methods reveal tRNA identity element divergence between Proteobacteria and Cyanobacteria. Biochimie, 89, 1276–1288. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
