

Structure-Guided Design of G-Protein-Coupled Receptor Polypharmacology
- PMID: 33904641
- PMCID: PMC8456950
- DOI: 10.1002/anie.202101478
Structure-Guided Design of G-Protein-Coupled Receptor Polypharmacology
Abstract
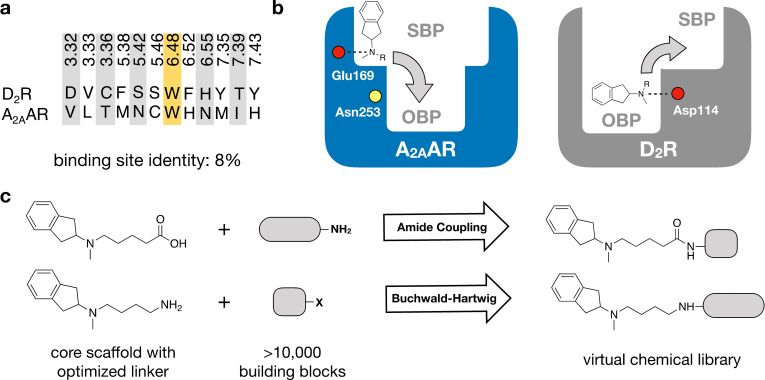
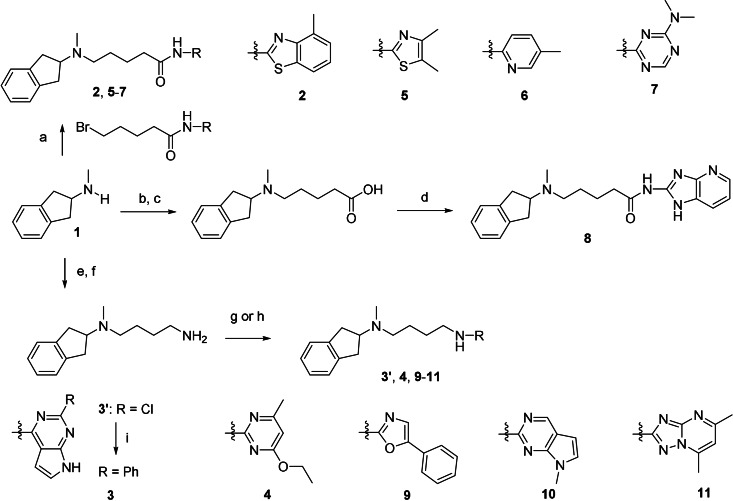
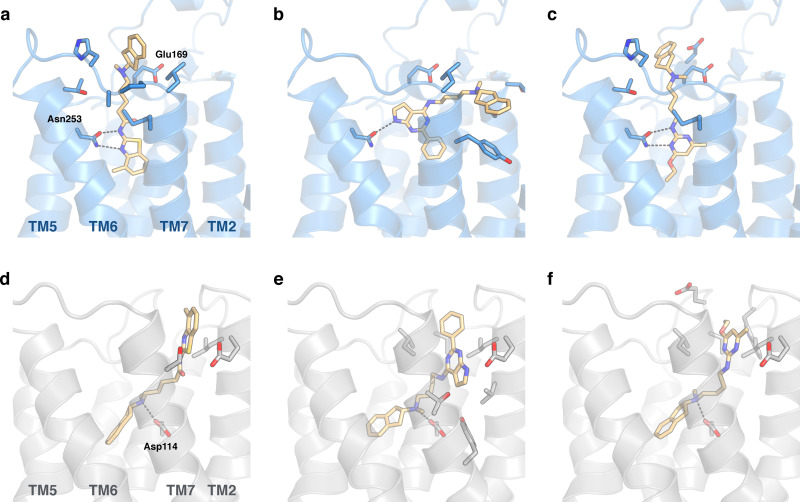
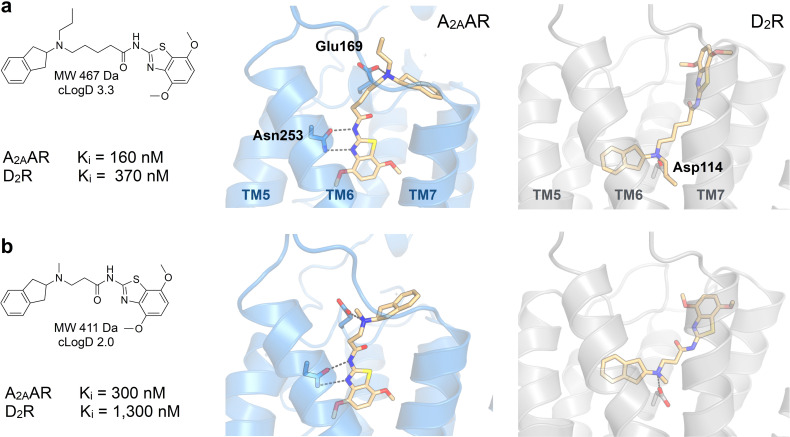
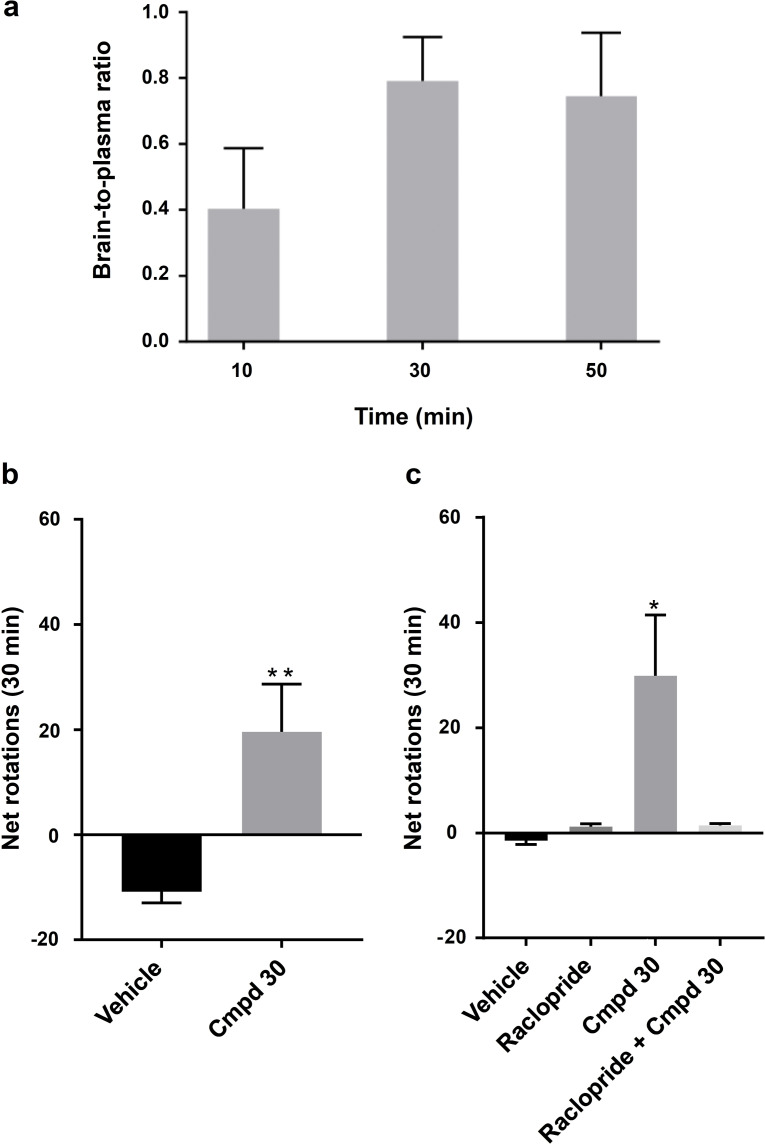
Many diseases are polygenic and can only be treated efficiently with drugs that modulate multiple targets. However, rational design of compounds with multi-target profiles is rarely pursued because it is considered too difficult, in particular if the drug must enter the central nervous system. Here, a structure-based strategy to identify dual-target ligands of G-protein-coupled receptors is presented. We use this approach to design compounds that both antagonize the A2A adenosine receptor and activate the D2 dopamine receptor, which have excellent potential as antiparkinson drugs. Atomic resolution models of the receptors guided generation of a chemical library with compounds designed to occupy orthosteric and secondary binding pockets in both targets. Structure-based virtual screens identified ten compounds, of which three had affinity for both targets. One of these scaffolds was optimized to nanomolar dual-target activity and showed the predicted pharmacodynamic effect in a rat model of Parkinsonism.
Keywords: Parkinson's disease; drug design; polypharmacology; receptors; virtual screening.
© 2021 The Authors. Angewandte Chemie International Edition published by Wiley-VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Charvin D., Medori R., Hauser R. A., Rascol O., Nat. Rev. Drug Discovery 2018, 17, 804–822. - PubMed
-
- Wong M. L., Licinio J., Nat. Rev. Drug Discovery 2004, 3, 136–151. - PubMed
-
- Roth B. L., Sheffer D. J., Kroeze W. K., Nat. Rev. Drug Discovery 2004, 3, 353–359. - PubMed
-
- Cavalli A., Bolognesi M. L., Mìnarini A., Rosini M., Tumiatti V., Recanatini M., Melchiorre C., J. Med. Chem. 2008, 51, 347–372. - PubMed
-
- Hopkins A. L., Nat. Chem. Biol. 2008, 4, 682–690. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
