

Frataxin deficiency promotes endothelial senescence in pulmonary hypertension
- PMID: 33905372
- PMCID: PMC8159699
- DOI: 10.1172/JCI136459
Frataxin deficiency promotes endothelial senescence in pulmonary hypertension
Abstract
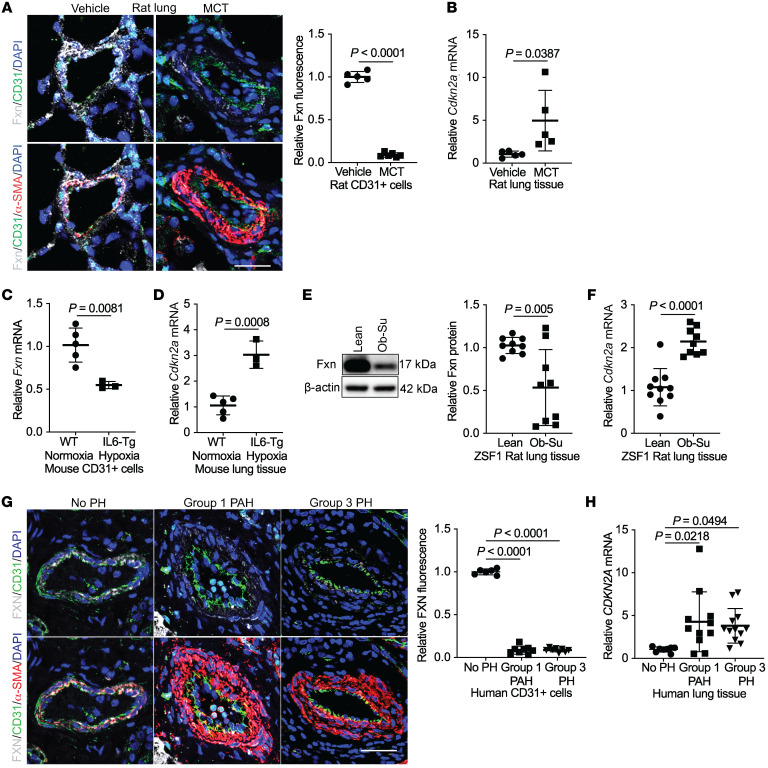
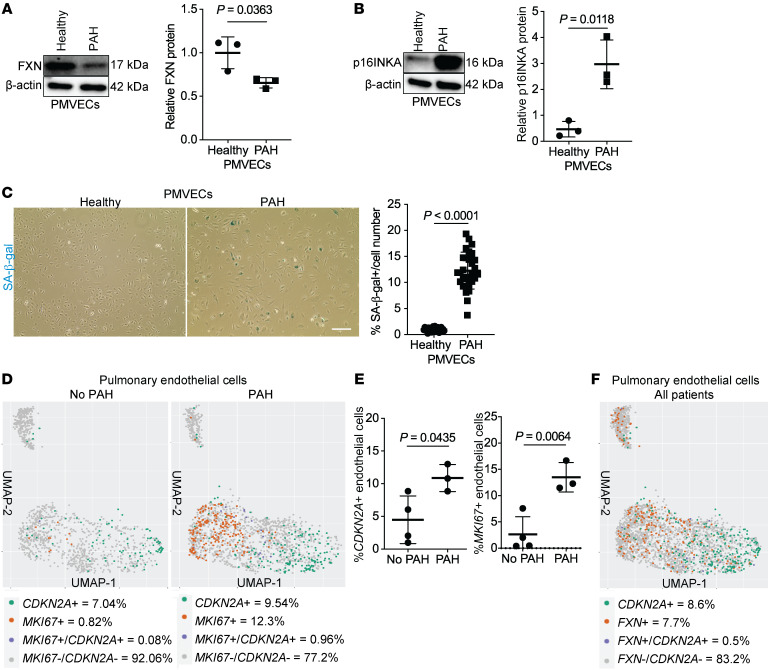
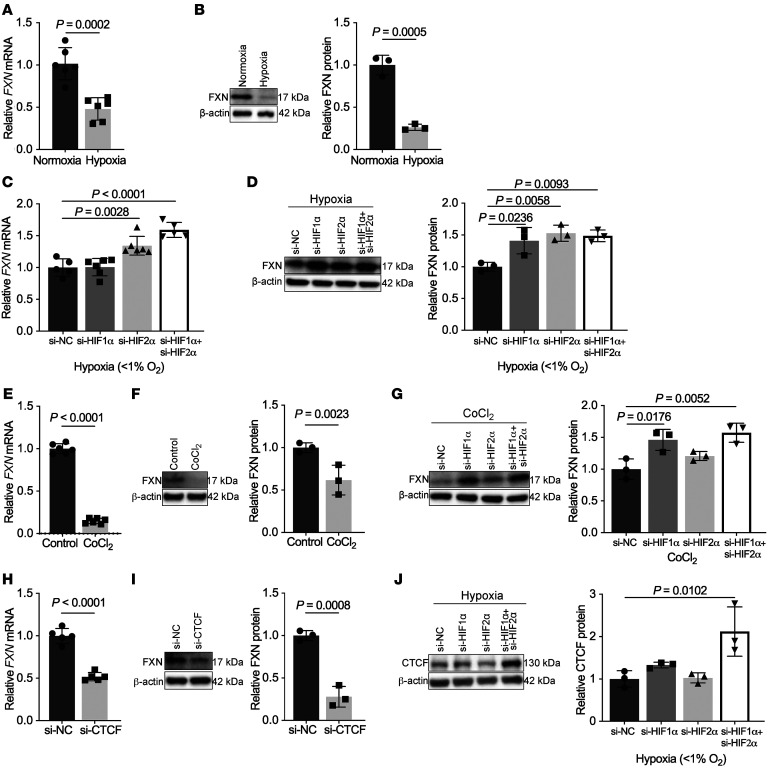
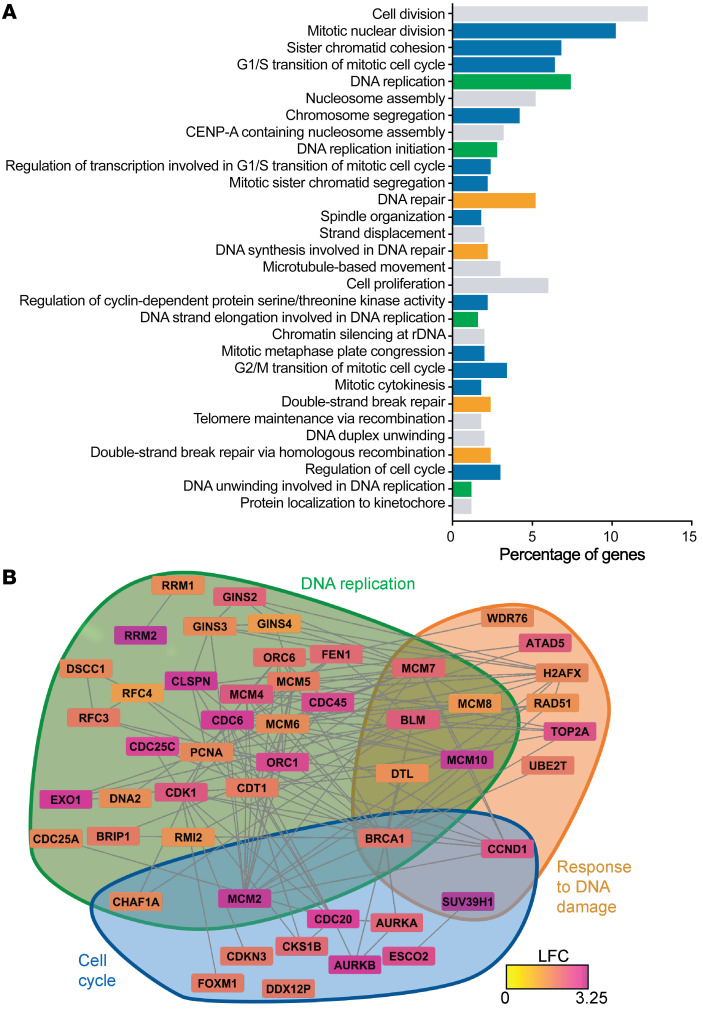
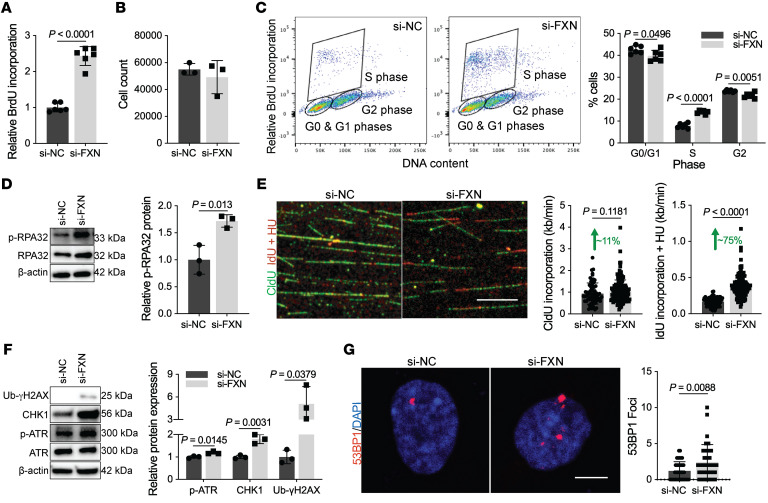
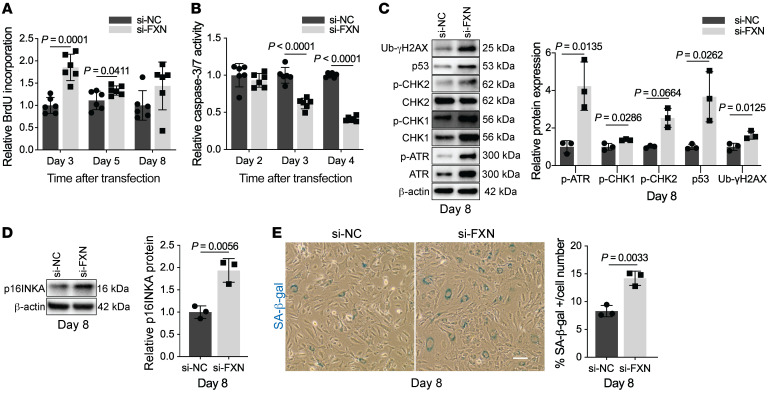
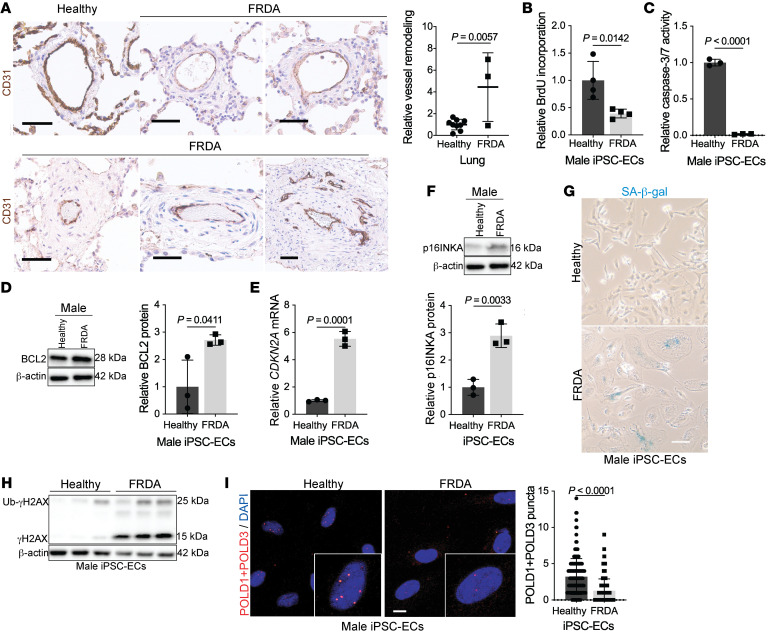
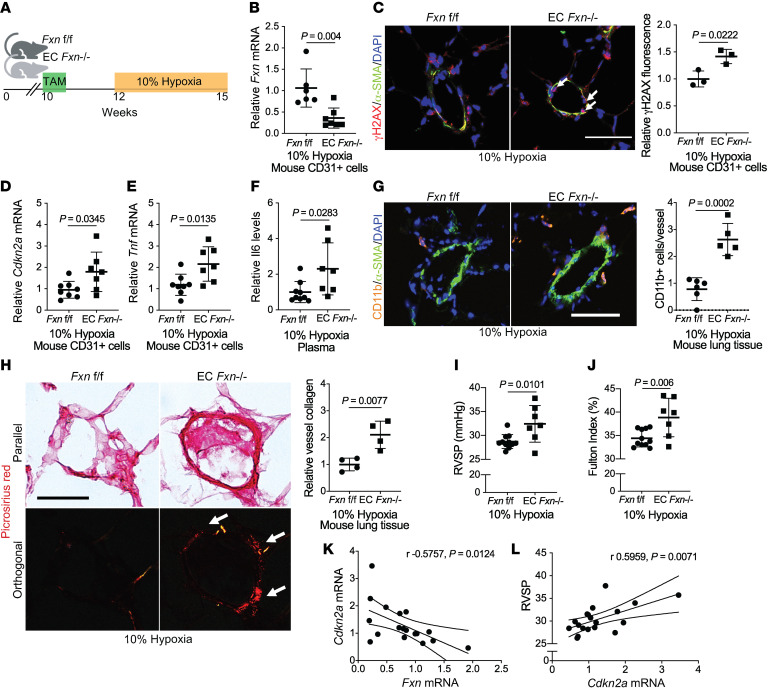
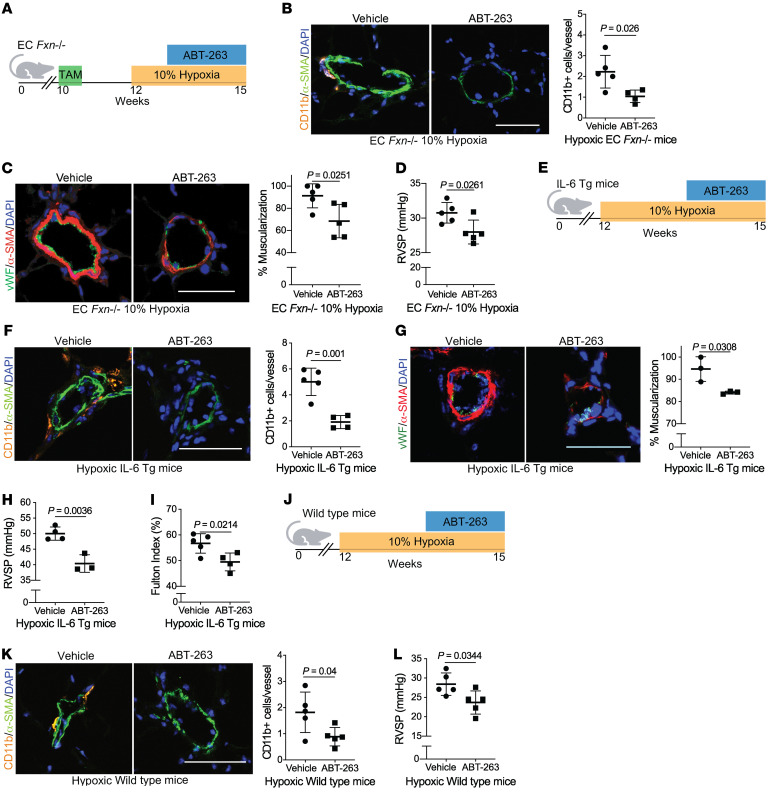
The dynamic regulation of endothelial pathophenotypes in pulmonary hypertension (PH) remains undefined. Cellular senescence is linked to PH with intracardiac shunts; however, its regulation across PH subtypes is unknown. Since endothelial deficiency of iron-sulfur (Fe-S) clusters is pathogenic in PH, we hypothesized that a Fe-S biogenesis protein, frataxin (FXN), controls endothelial senescence. An endothelial subpopulation in rodent and patient lungs across PH subtypes exhibited reduced FXN and elevated senescence. In vitro, hypoxic and inflammatory FXN deficiency abrogated activity of endothelial Fe-S-containing polymerases, promoting replication stress, DNA damage response, and senescence. This was also observed in stem cell-derived endothelial cells from Friedreich's ataxia (FRDA), a genetic disease of FXN deficiency, ataxia, and cardiomyopathy, often with PH. In vivo, FXN deficiency-dependent senescence drove vessel inflammation, remodeling, and PH, whereas pharmacologic removal of senescent cells in Fxn-deficient rodents ameliorated PH. These data offer a model of endothelial biology in PH, where FXN deficiency generates a senescent endothelial subpopulation, promoting vascular inflammatory and proliferative signals in other cells to drive disease. These findings also establish an endothelial etiology for PH in FRDA and left heart disease and support therapeutic development of senolytic drugs, reversing effects of Fe-S deficiency across PH subtypes.
Keywords: Cardiovascular disease; Endothelial cells; Hypertension; Pulmonology; Vascular Biology.
Conflict of interest statement
Figures
Comment in
-
Frataxin and endothelial cell senescence in pulmonary hypertension.J Clin Invest. 2021 Jun 1;131(11):e149721. doi: 10.1172/JCI149721. J Clin Invest. 2021. PMID: 34060487 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
- R00 HL135258/HL/NHLBI NIH HHS/United States
- U01 HL145550/HL/NHLBI NIH HHS/United States
- R01 HL138437/HL/NHLBI NIH HHS/United States
- P30 CA014051/CA/NCI NIH HHS/United States
- K99 HL135258/HL/NHLBI NIH HHS/United States
- R01 HL122596/HL/NHLBI NIH HHS/United States
- UH3 TR002073/TR/NCATS NIH HHS/United States
- R01 HL150638/HL/NHLBI NIH HHS/United States
- F30 HL139017/HL/NHLBI NIH HHS/United States
- S10 OD023684/OD/NIH HHS/United States
- R00 AG049126/AG/NIA NIH HHS/United States
- R01 HL130261/HL/NHLBI NIH HHS/United States
- UH2 TR002073/TR/NCATS NIH HHS/United States
- R01 HL124021/HL/NHLBI NIH HHS/United States
- R01 HL113178/HL/NHLBI NIH HHS/United States
- T32 HL110849/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
