

Protection of the C. elegans germ cell genome depends on diverse DNA repair pathways during normal proliferation
- PMID: 33905417
- PMCID: PMC8078821
- DOI: 10.1371/journal.pone.0250291
Protection of the C. elegans germ cell genome depends on diverse DNA repair pathways during normal proliferation
Abstract
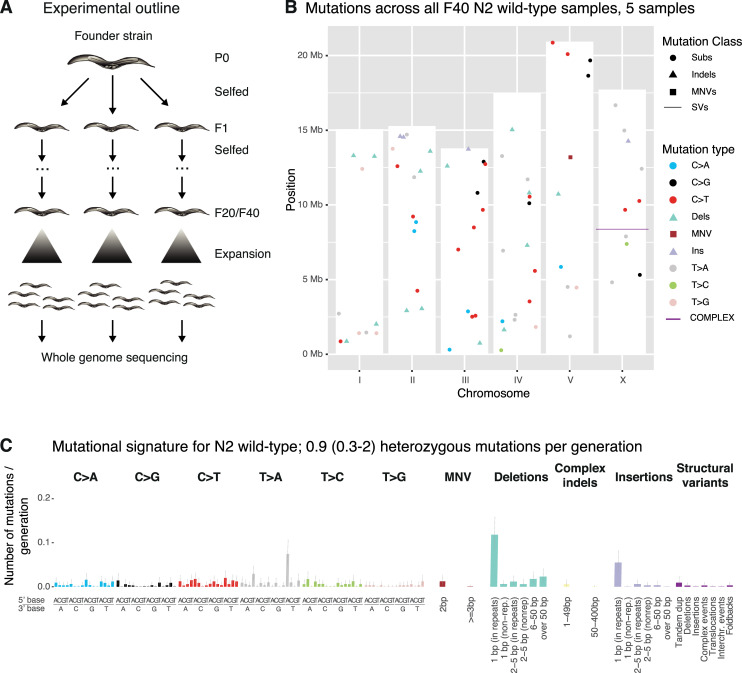
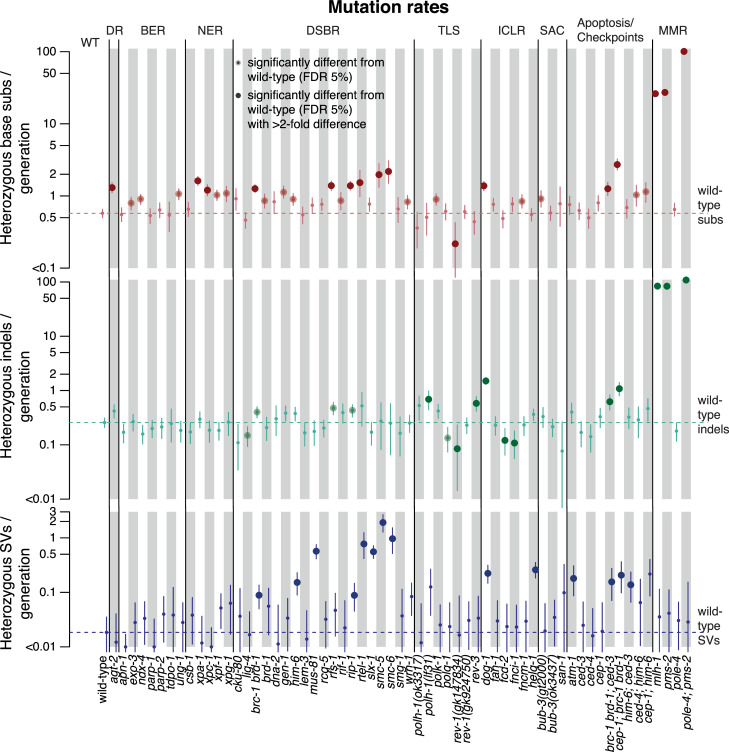
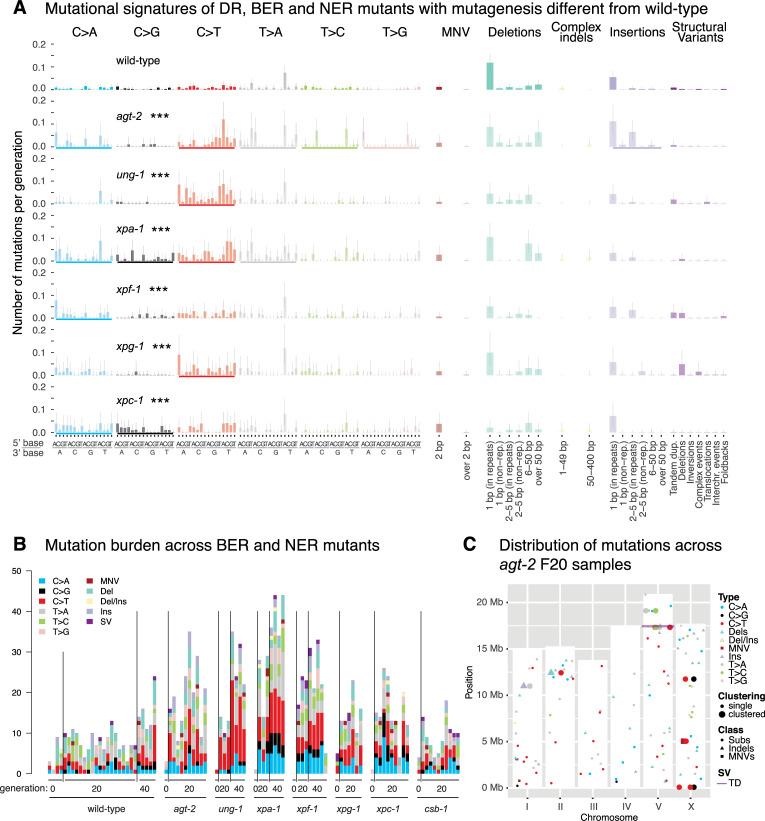
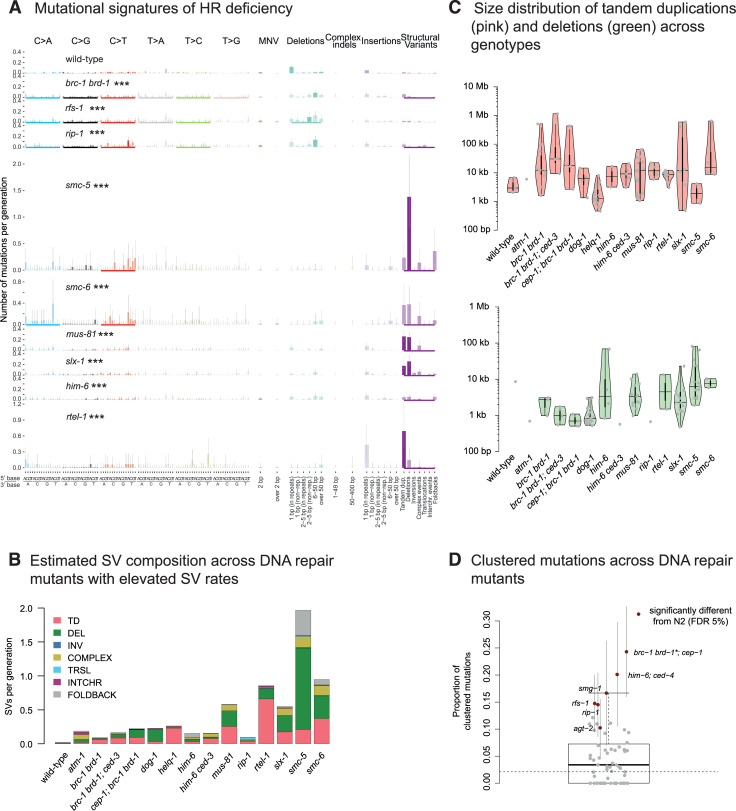
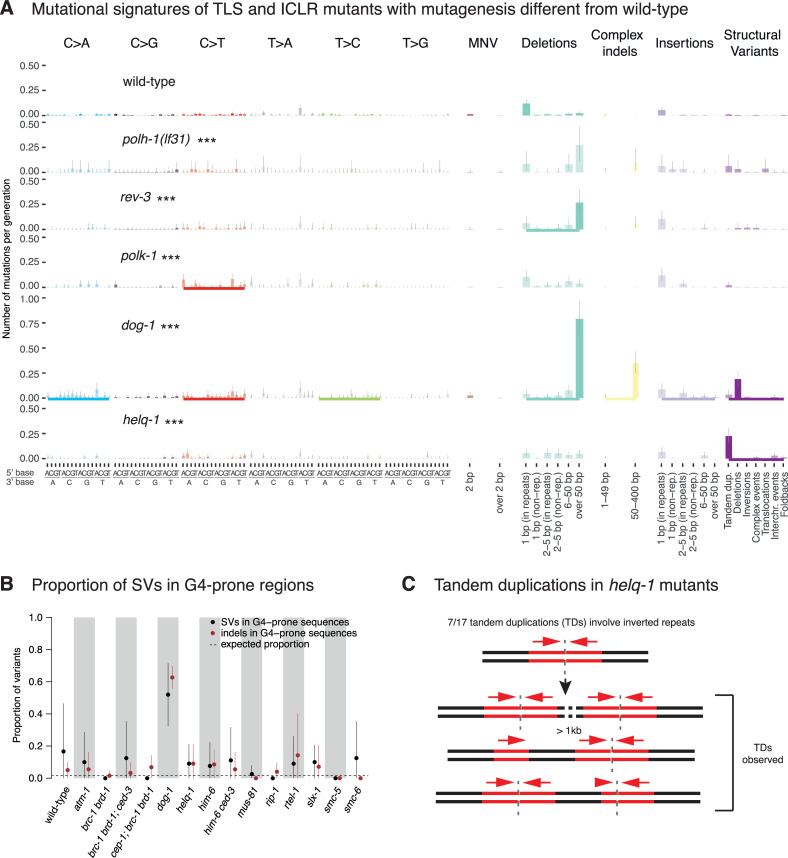
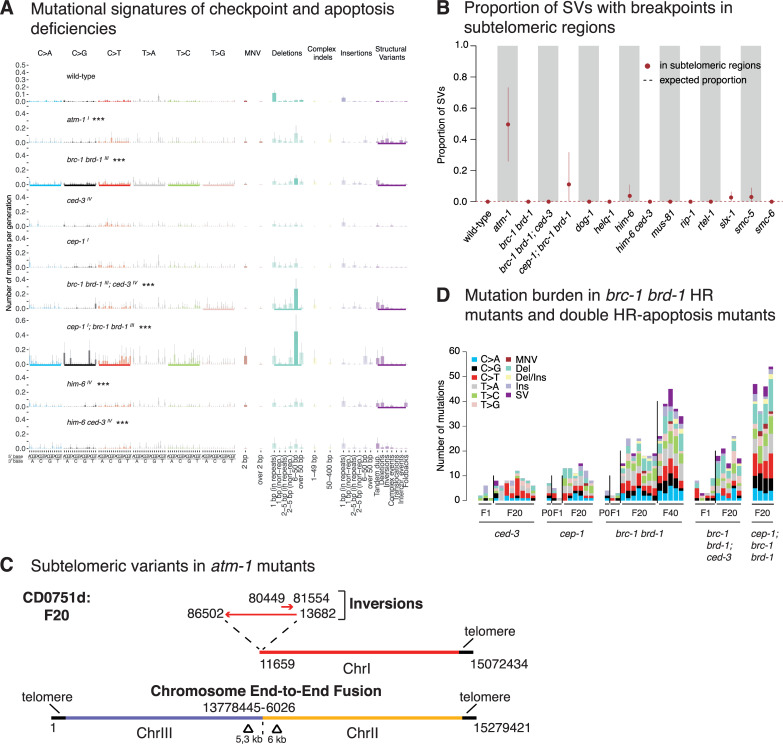
Maintaining genome integrity is particularly important in germ cells to ensure faithful transmission of genetic information across generations. Here we systematically describe germ cell mutagenesis in wild-type and 61 DNA repair mutants cultivated over multiple generations. ~44% of the DNA repair mutants analysed showed a >2-fold increased mutagenesis with a broad spectrum of mutational outcomes. Nucleotide excision repair deficiency led to higher base substitution rates, whereas polh-1(Polη) and rev-3(Polζ) translesion synthesis polymerase mutants resulted in 50-400 bp deletions. Signatures associated with defective homologous recombination fall into two classes: 1) brc-1/BRCA1 and rad-51/RAD51 paralog mutants showed increased mutations across all mutation classes, 2) mus-81/MUS81 and slx-1/SLX1 nuclease, and him-6/BLM, helq-1/HELQ or rtel-1/RTEL1 helicase mutants primarily accumulated structural variants. Repetitive and G-quadruplex sequence-containing loci were more frequently mutated in specific DNA repair backgrounds. Tandem duplications embedded in inverted repeats were observed in helq-1 helicase mutants, and a unique pattern of 'translocations' involving homeologous sequences occurred in rip-1 recombination mutants. atm-1/ATM checkpoint mutants harboured structural variants specifically enriched in subtelomeric regions. Interestingly, locally clustered mutagenesis was only observed for combined brc-1 and cep-1/p53 deficiency. Our study provides a global view of how different DNA repair pathways contribute to prevent germ cell mutagenesis.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
