

Single-cell sequencing of human white adipose tissue identifies new cell states in health and obesity
- PMID: 33907320
- PMCID: PMC8102391
- DOI: 10.1038/s41590-021-00922-4
Single-cell sequencing of human white adipose tissue identifies new cell states in health and obesity
Abstract
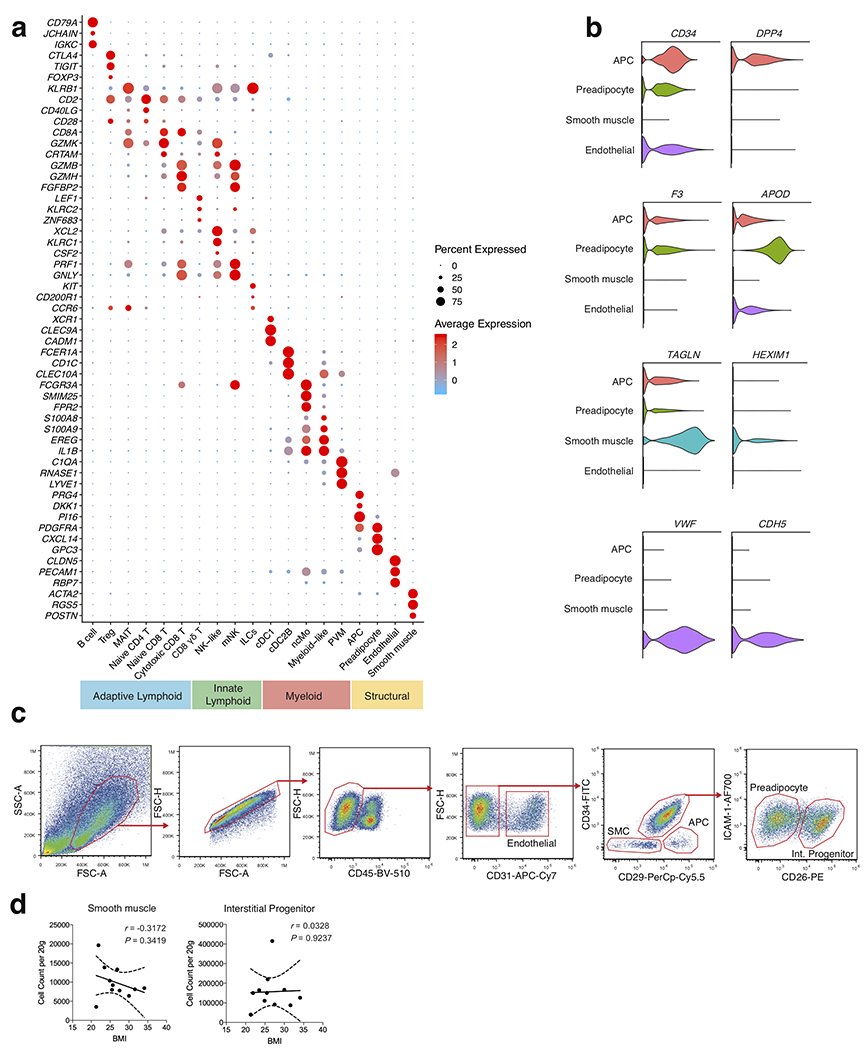
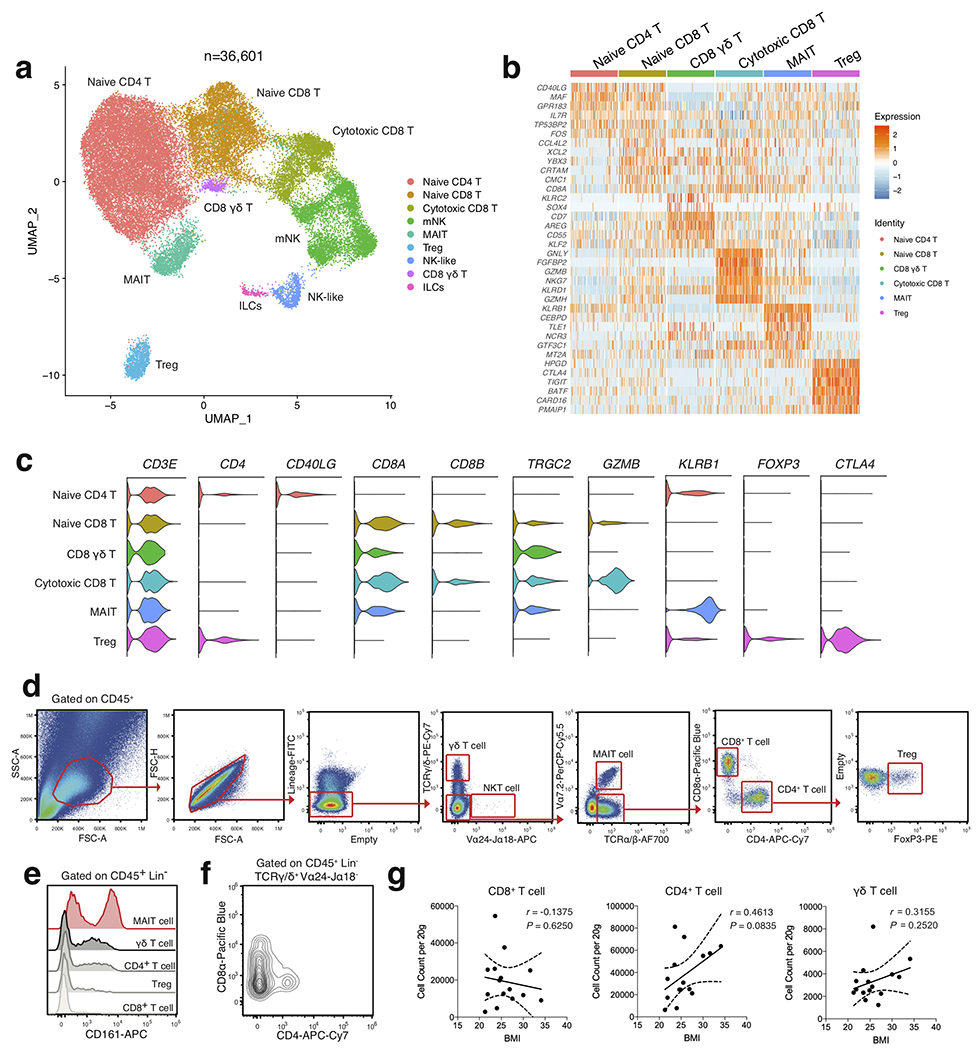
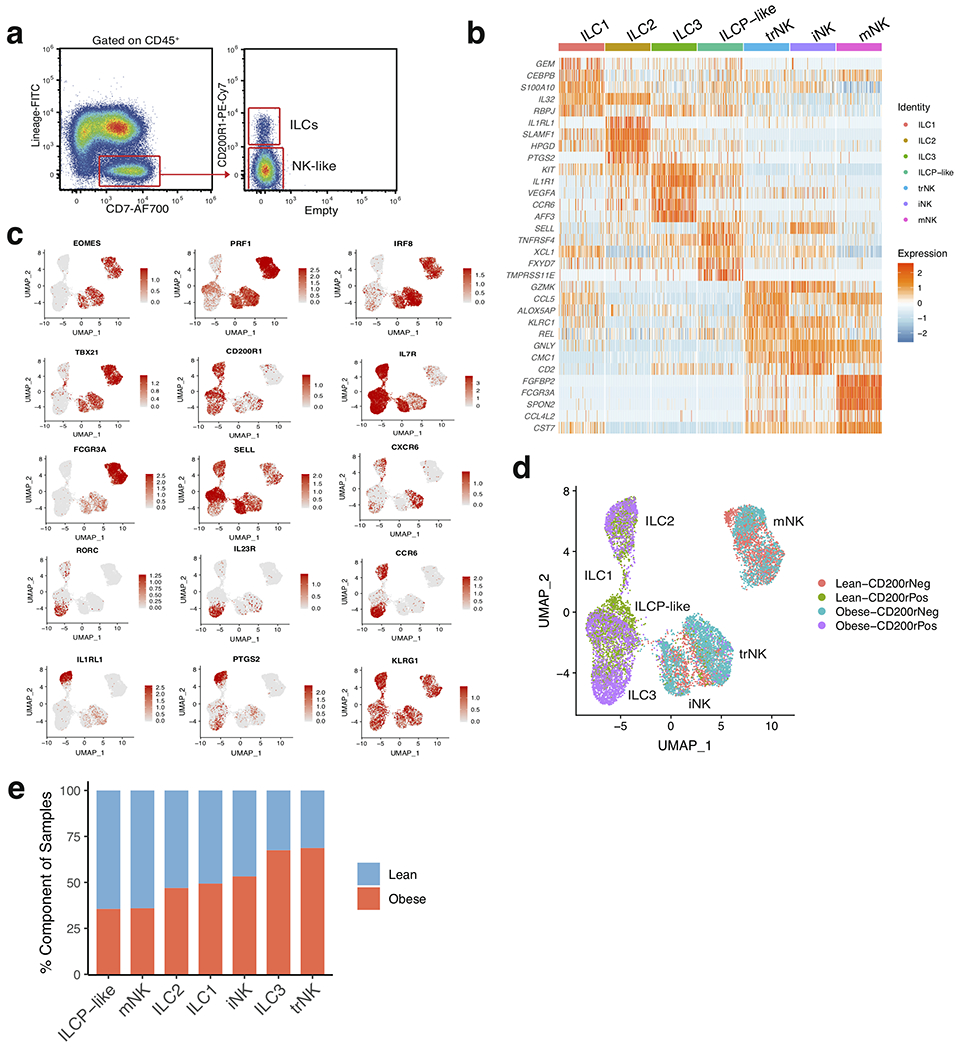
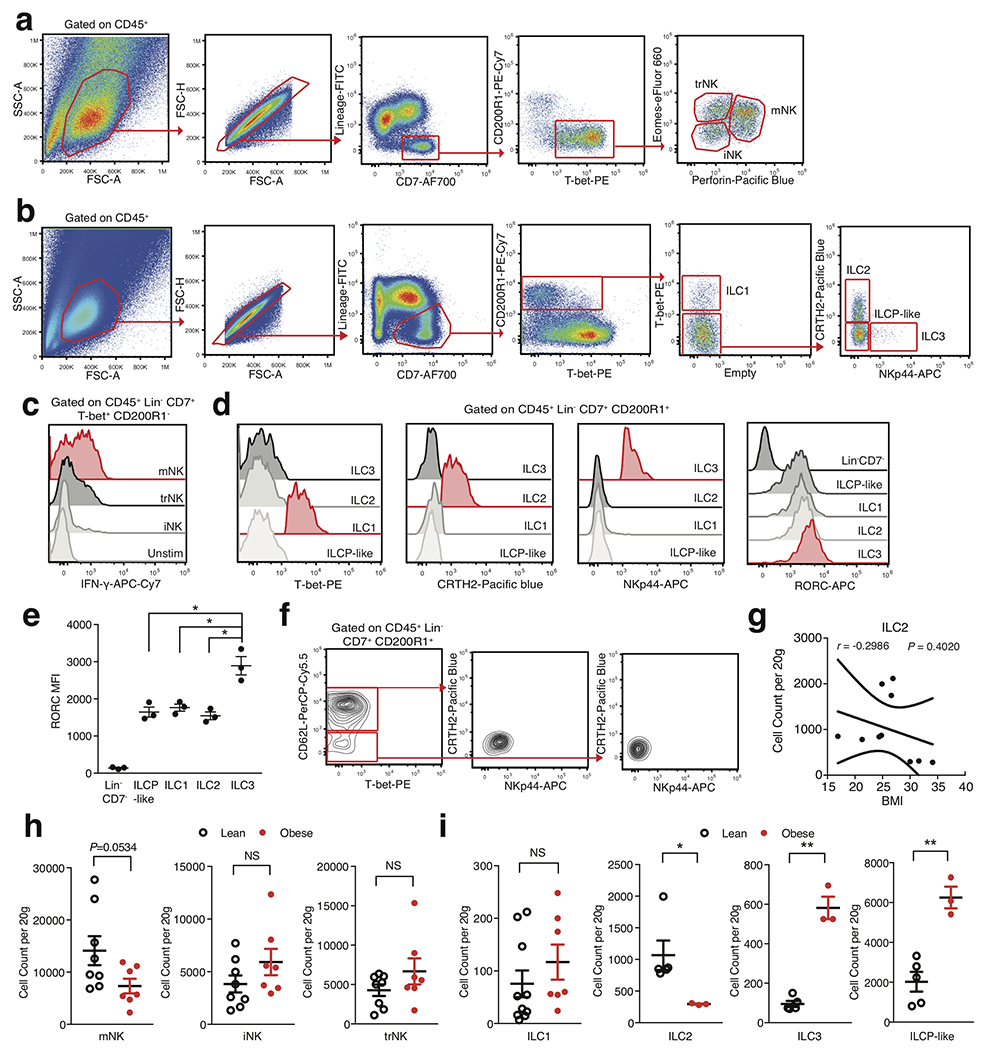
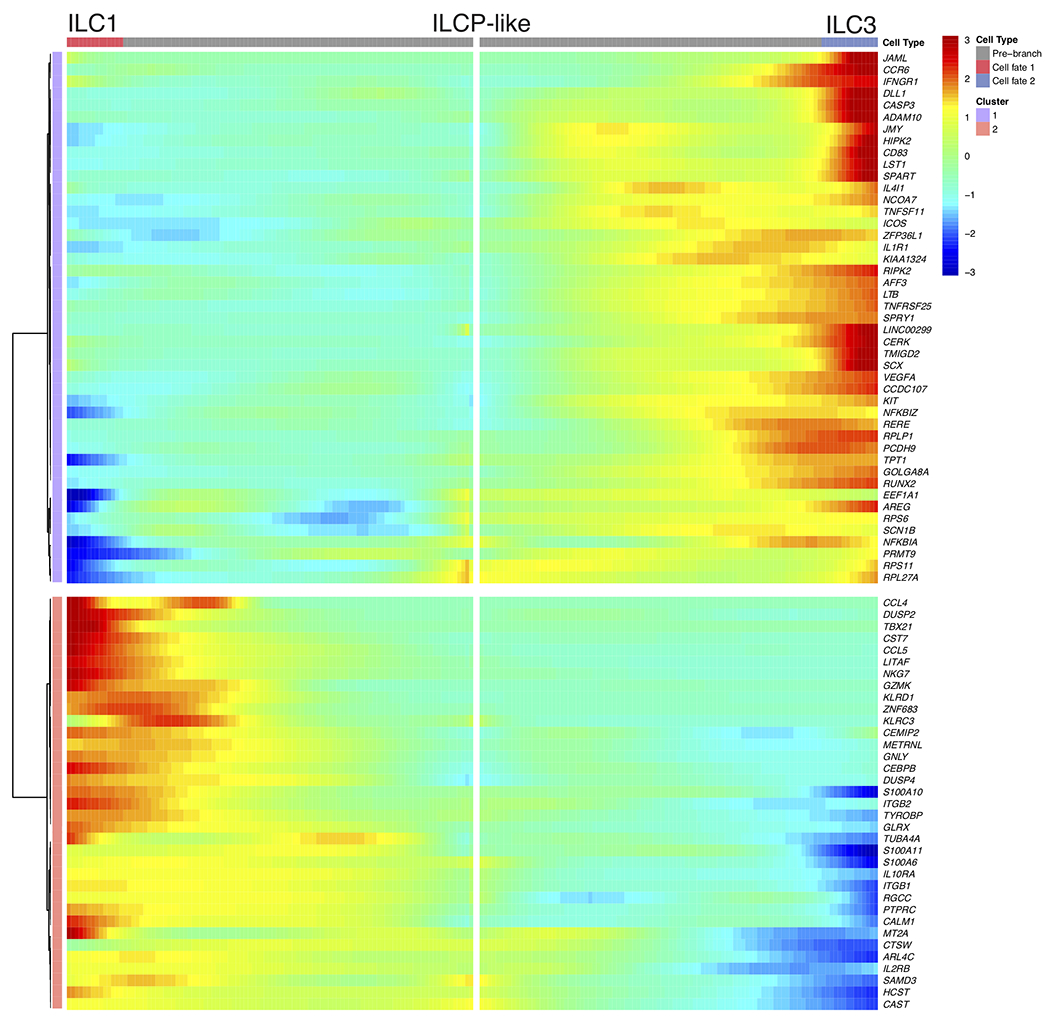
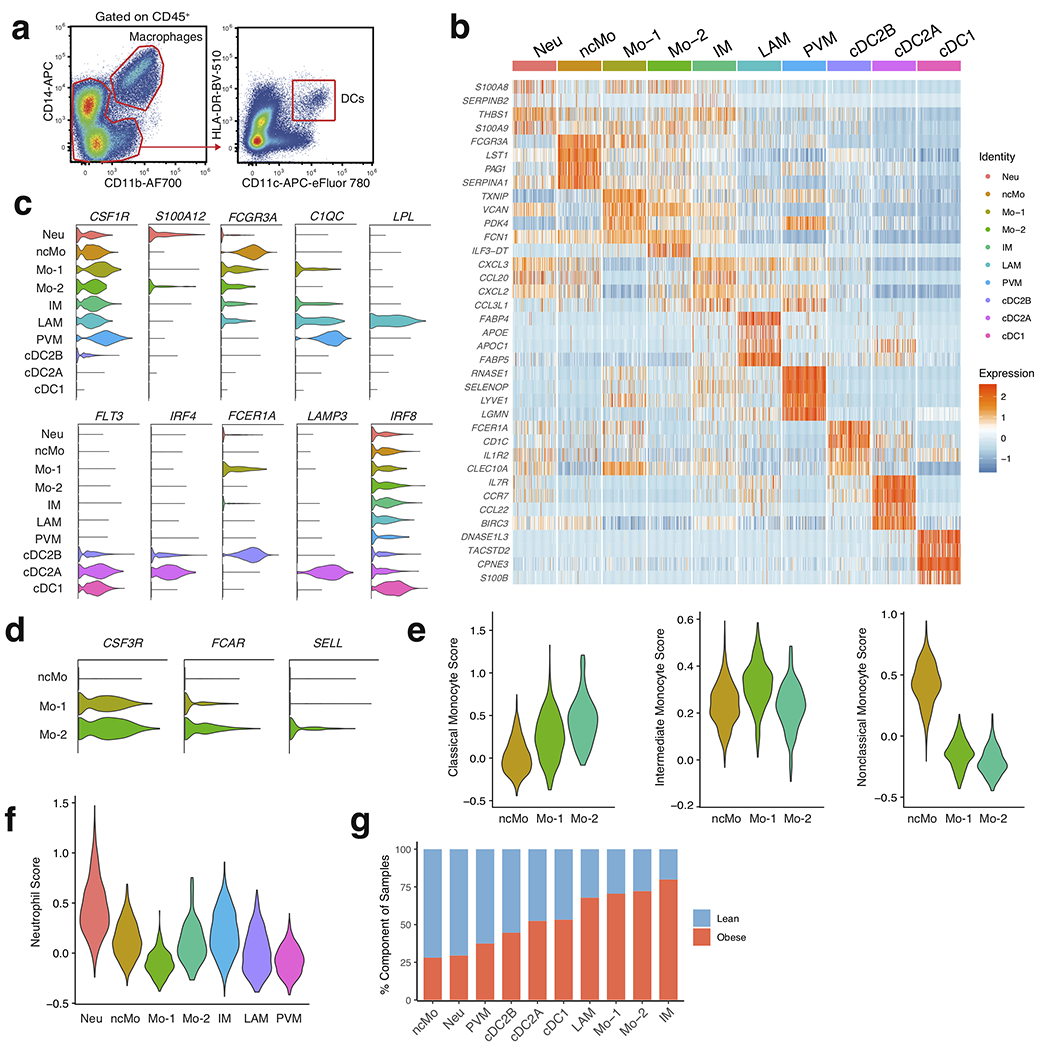
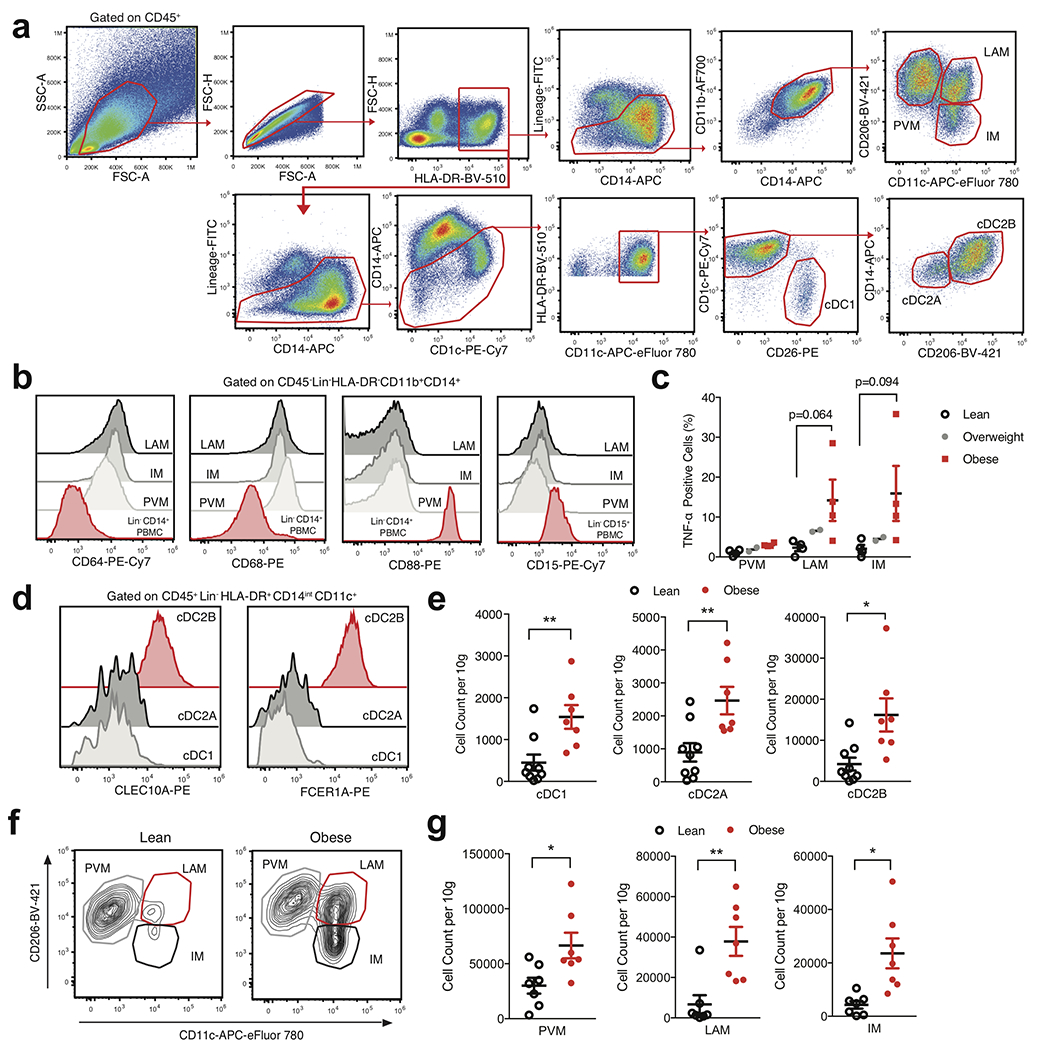
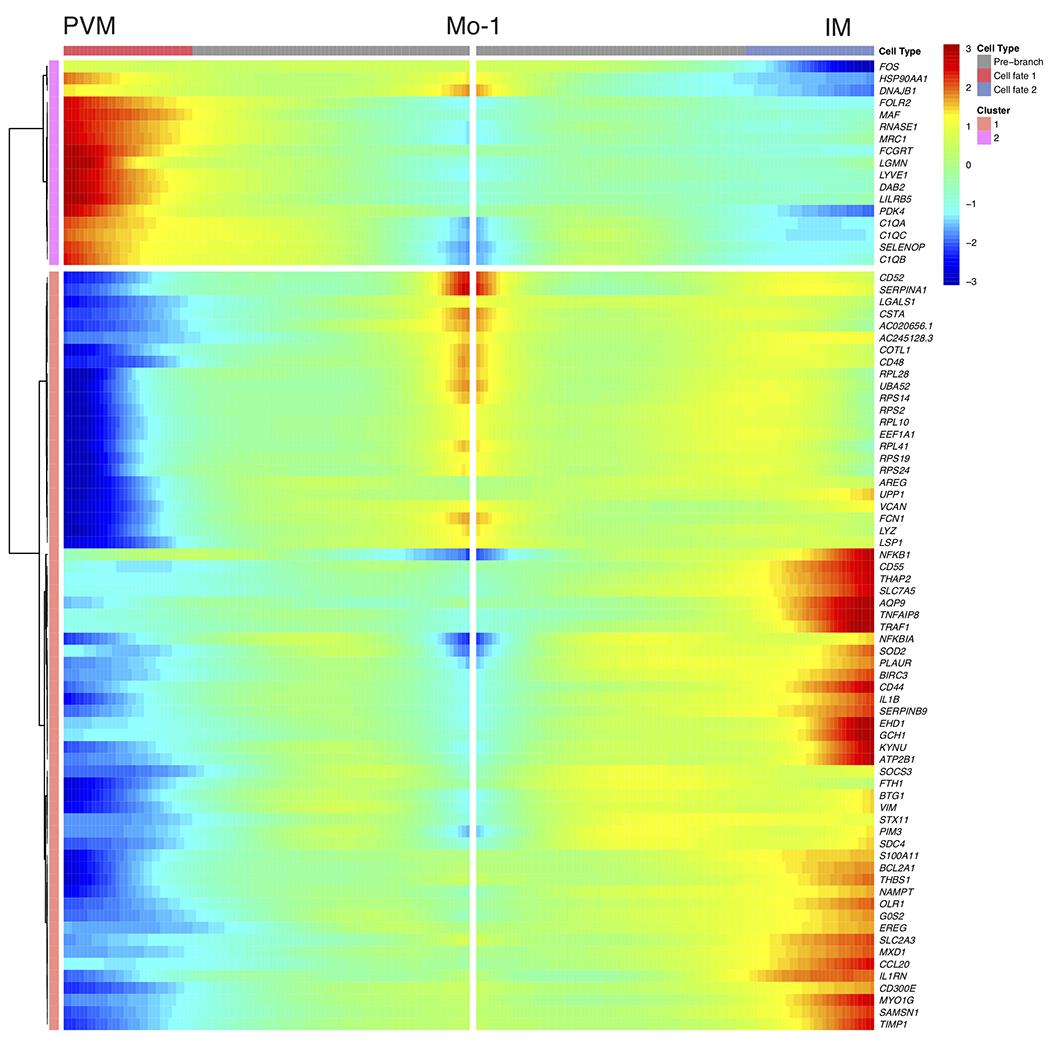
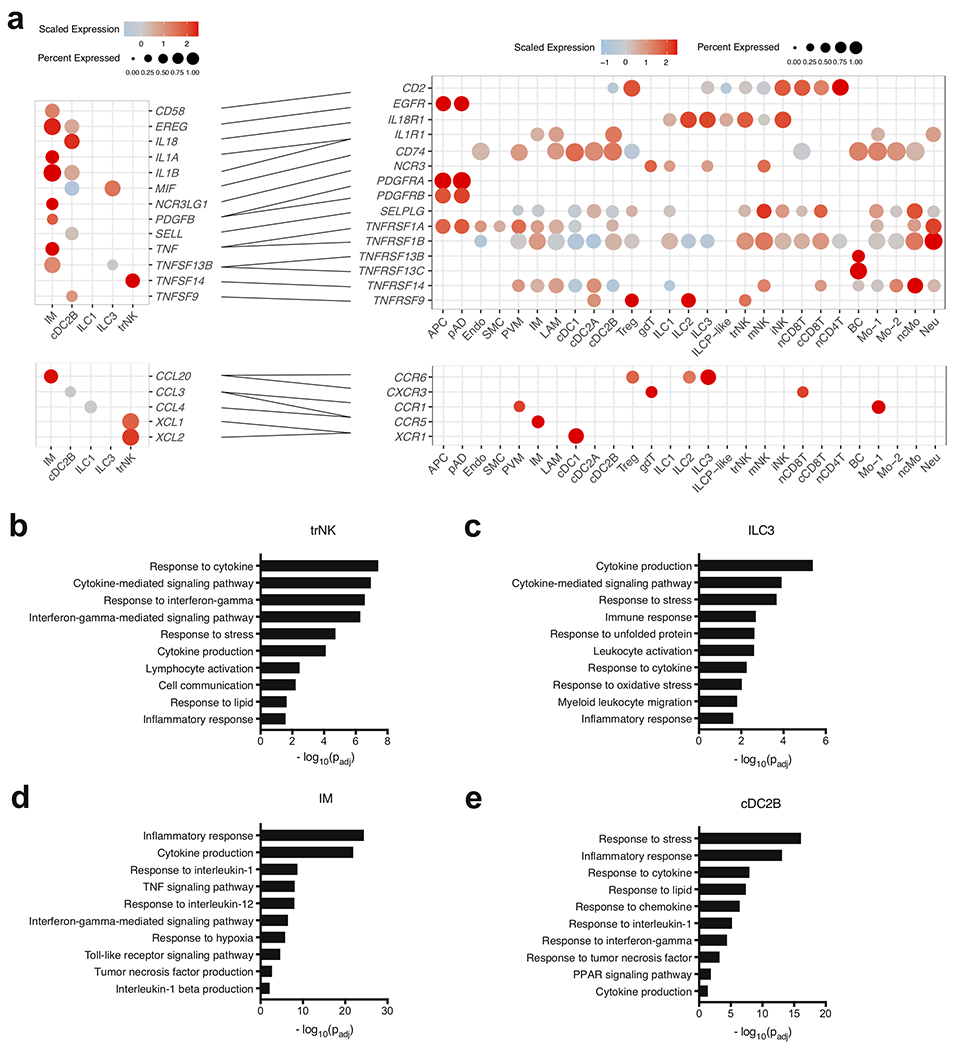
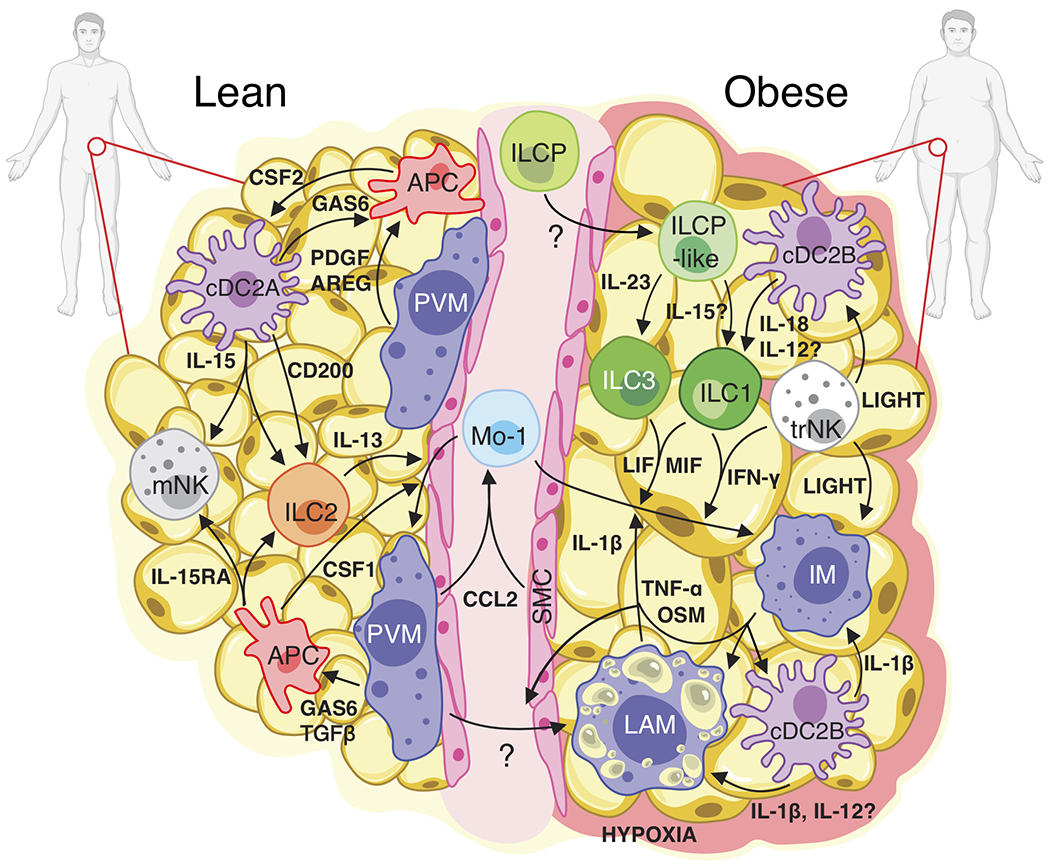
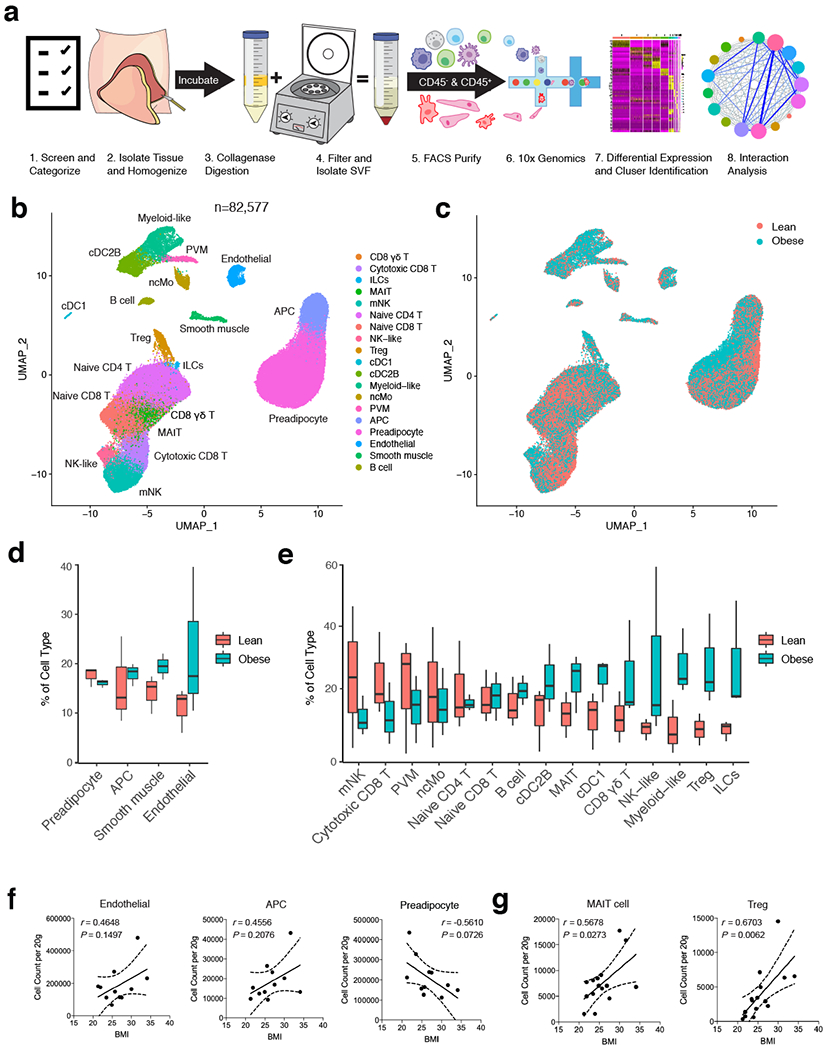
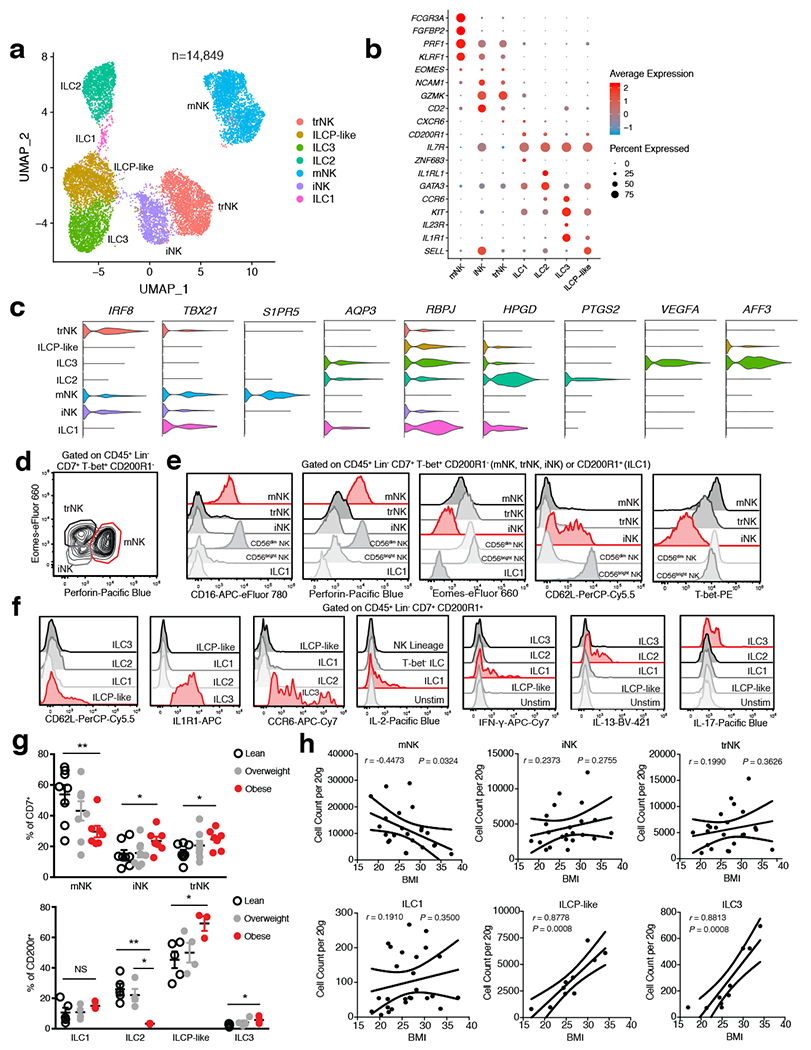
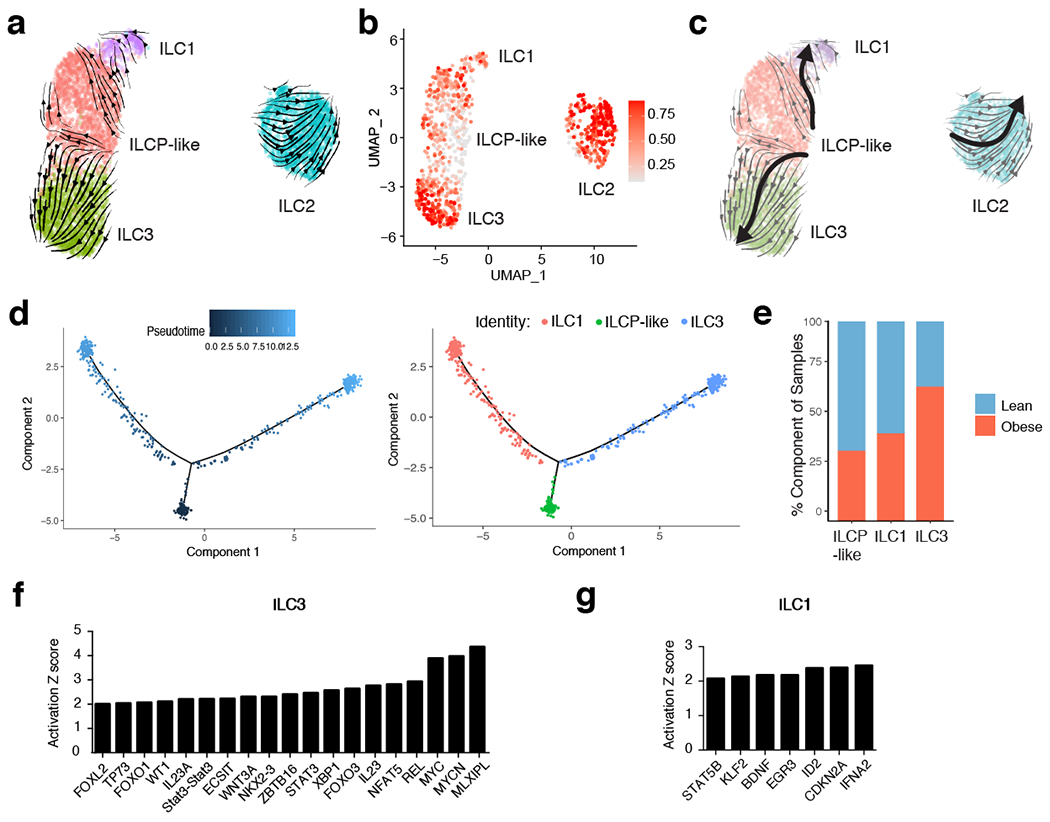
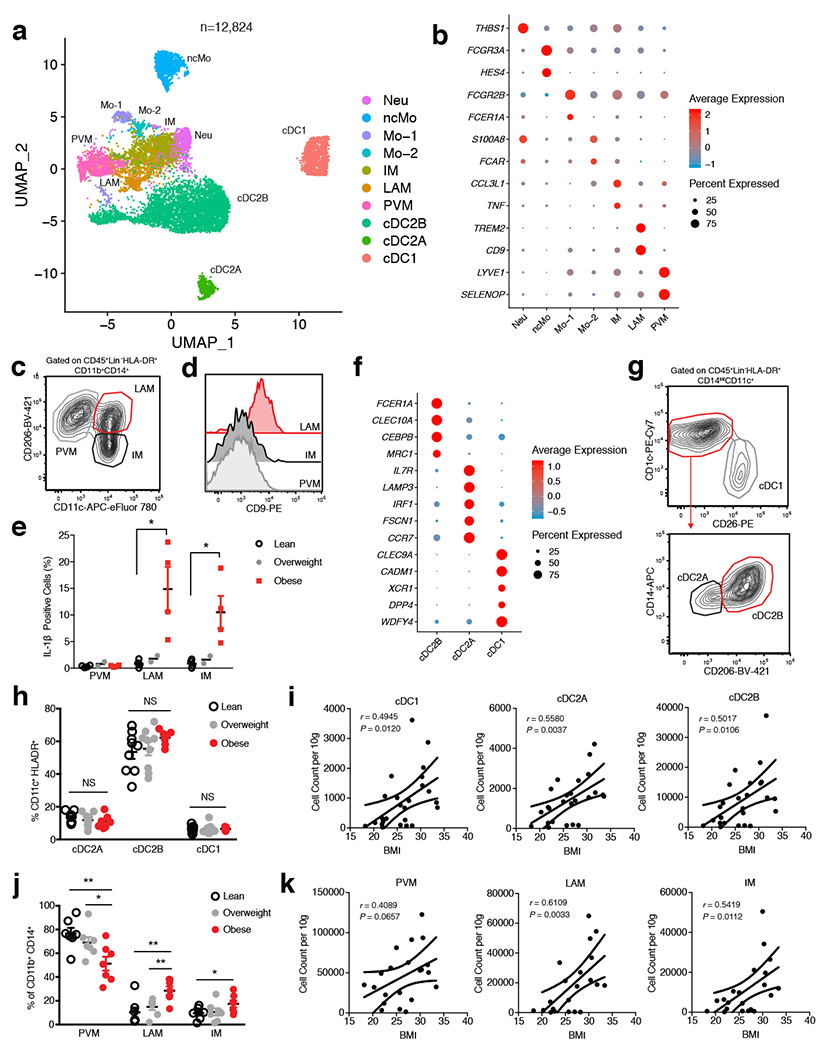
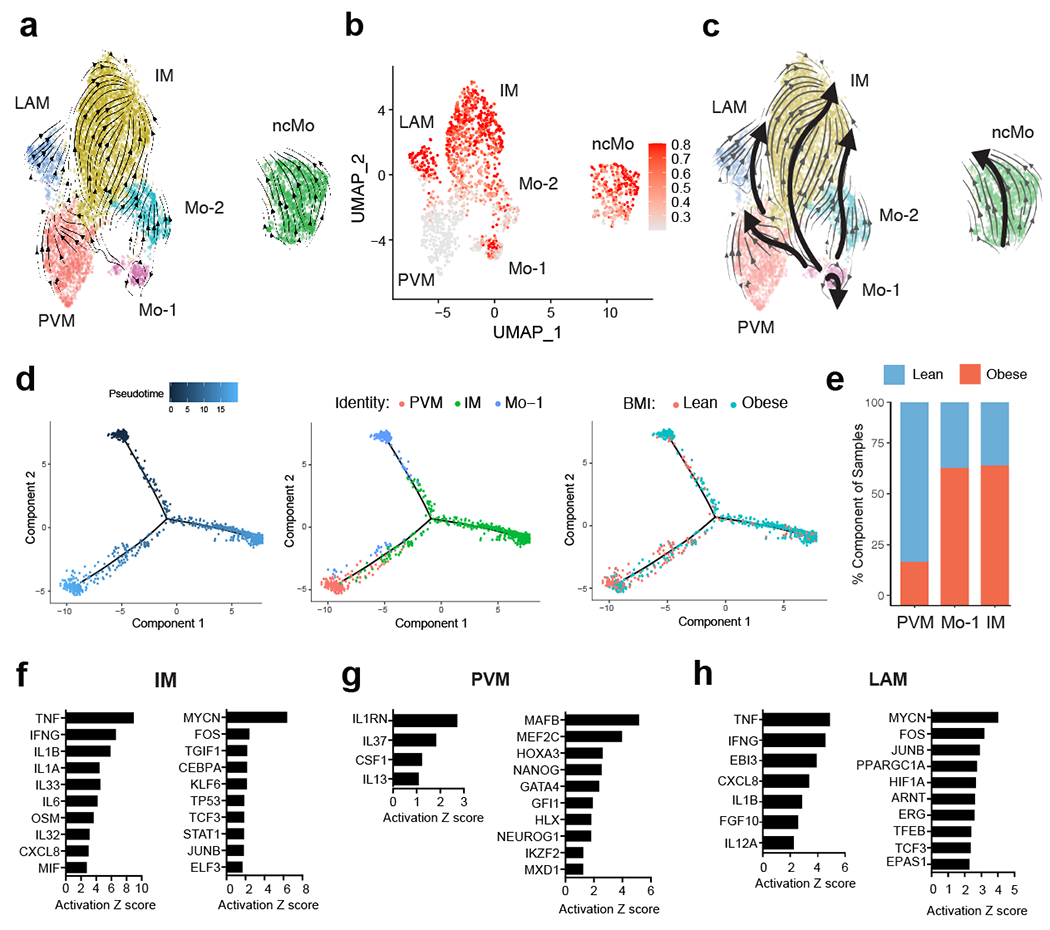
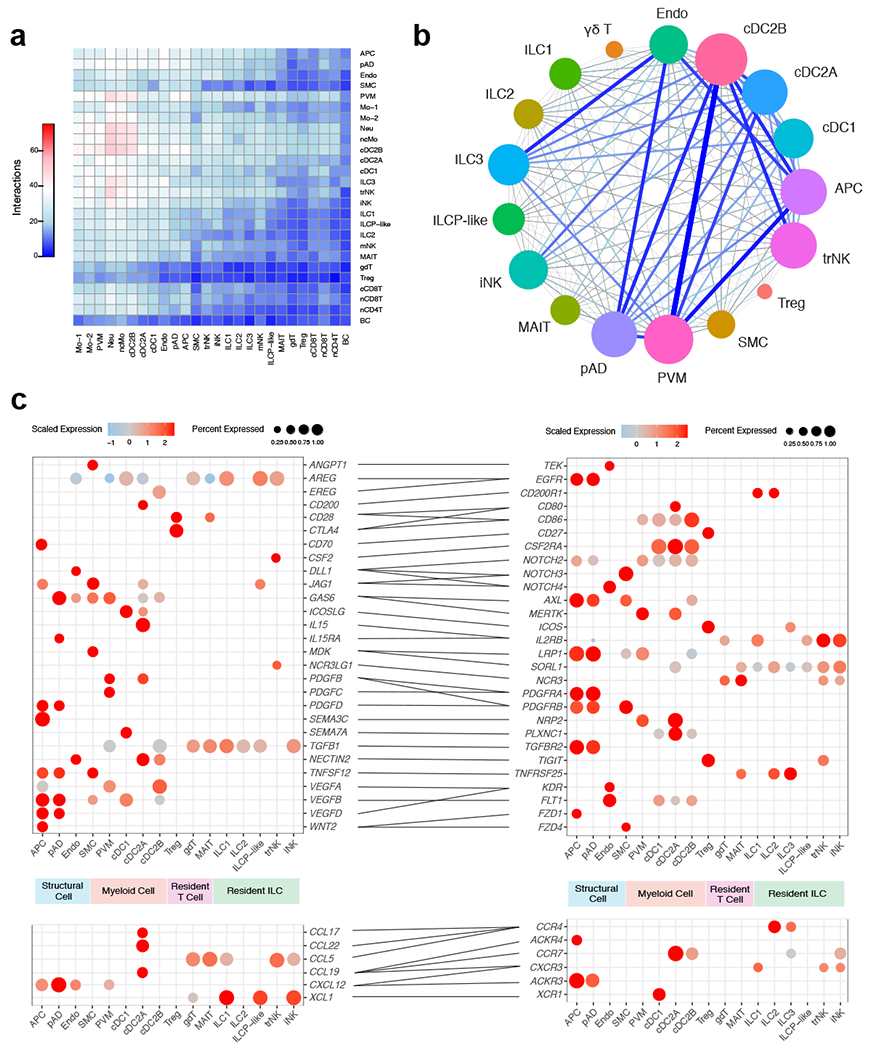
White adipose tissue (WAT) is an essential regulator of energy storage and systemic metabolic homeostasis. Regulatory networks consisting of immune and structural cells are necessary to maintain WAT metabolism, which can become impaired during obesity in mammals. Using single-cell transcriptomics and flow cytometry, we unveil a large-scale comprehensive cellular census of the stromal vascular fraction of healthy lean and obese human WAT. We report new subsets and developmental trajectories of adipose-resident innate lymphoid cells, dendritic cells and monocyte-derived macrophage populations that accumulate in obese WAT. Analysis of cell-cell ligand-receptor interactions and obesity-enriched signaling pathways revealed a switch from immunoregulatory mechanisms in lean WAT to inflammatory networks in obese WAT. These results provide a detailed and unbiased cellular landscape of homeostatic and inflammatory circuits in healthy human WAT.
Conflict of interest statement
Competing Interests Statement
The authors do not have any competing interests to declare.
Figures
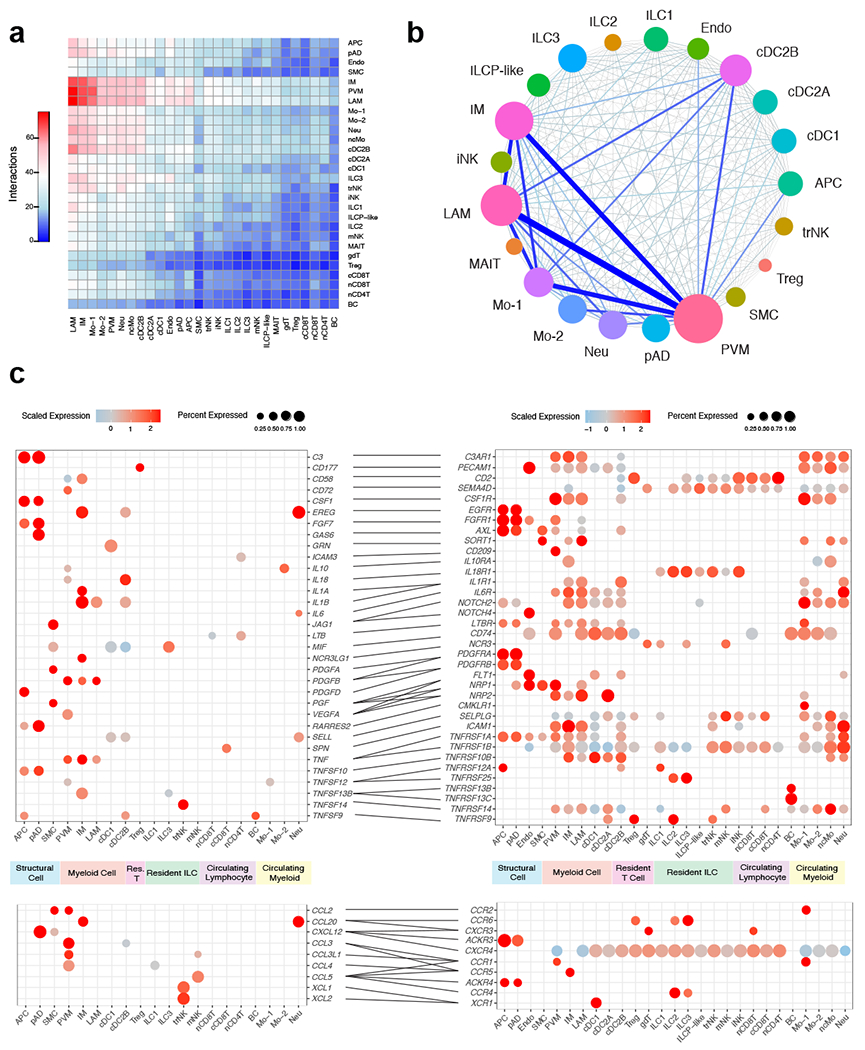
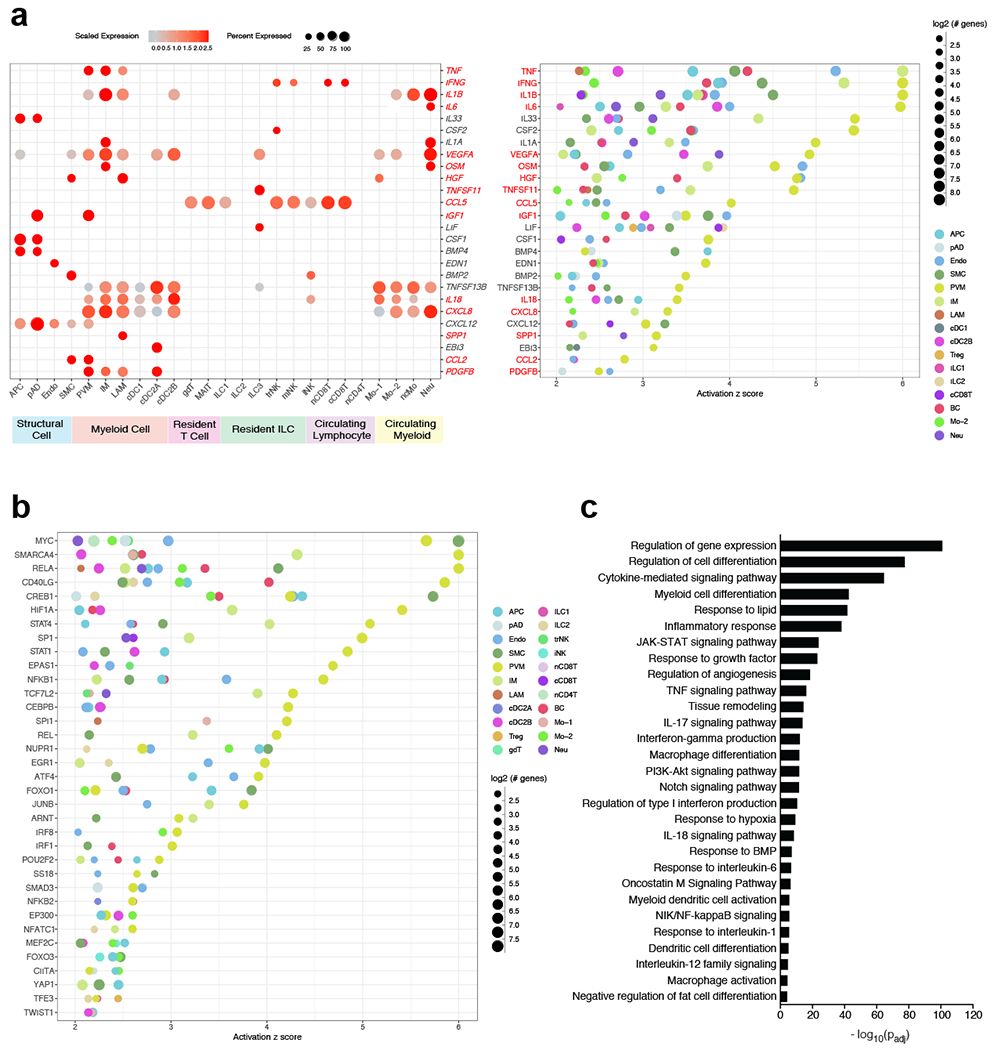
References
-
- Christ A, Lauterbach M & Latz E Western Diet and the Immune System: An Inflammatory Connection. Immunity 51, 794–811 (2019). - PubMed
-
- Roden M & Shulman GI The integrative biology of type 2 diabetes. Nature 576, 51–60 (2019). - PubMed
-
- Lackey DE & Olefsky JM Regulation of metabolism by the innate immune system. Nat Rev Endocrinol 12, 15–28 (2016). - PubMed
-
- Pollack RM, Donath MY, LeRoith D & Leibowitz G Anti-inflammatory Agents in the Treatment of Diabetes and Its Vascular Complications. Dia Care 39, S244–S252 (2016). - PubMed
Additional Methods References:
-
- Wong KL et al. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 118, e16–e31 (2011). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
