

Structure, Assembly, and Function of Tripartite Efflux and Type 1 Secretion Systems in Gram-Negative Bacteria
- PMID: 33909410
- PMCID: PMC8277102
- DOI: 10.1021/acs.chemrev.1c00055
Structure, Assembly, and Function of Tripartite Efflux and Type 1 Secretion Systems in Gram-Negative Bacteria
Abstract
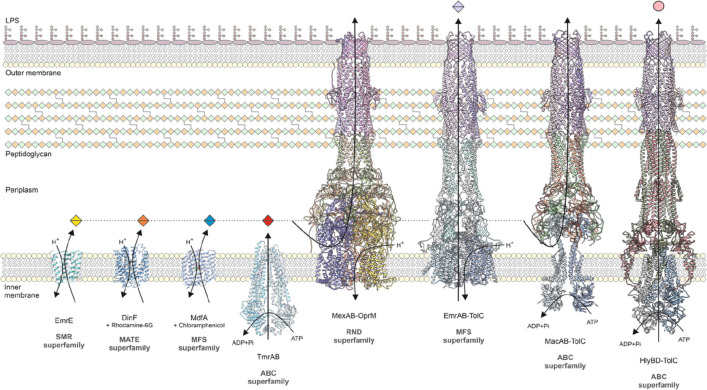
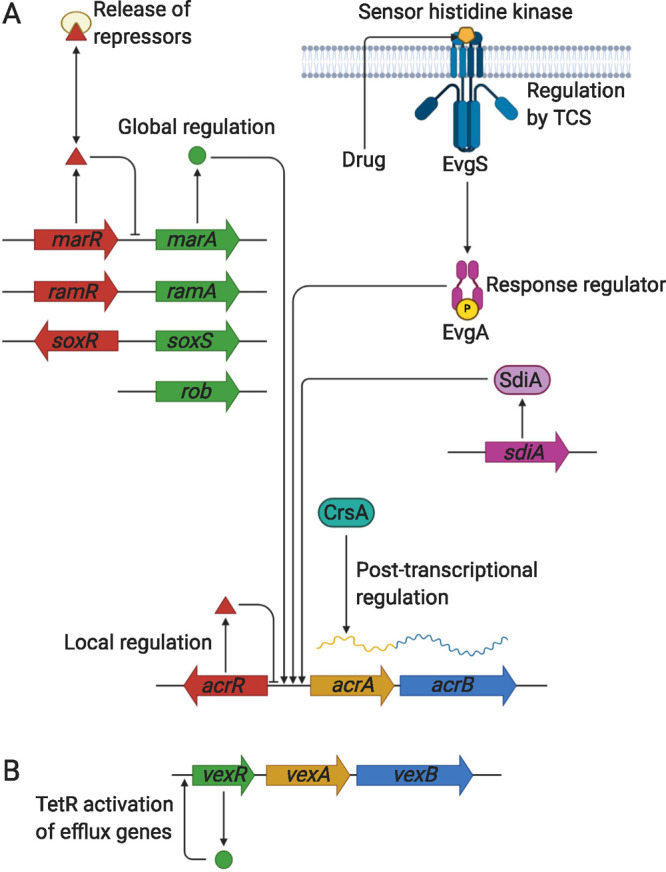
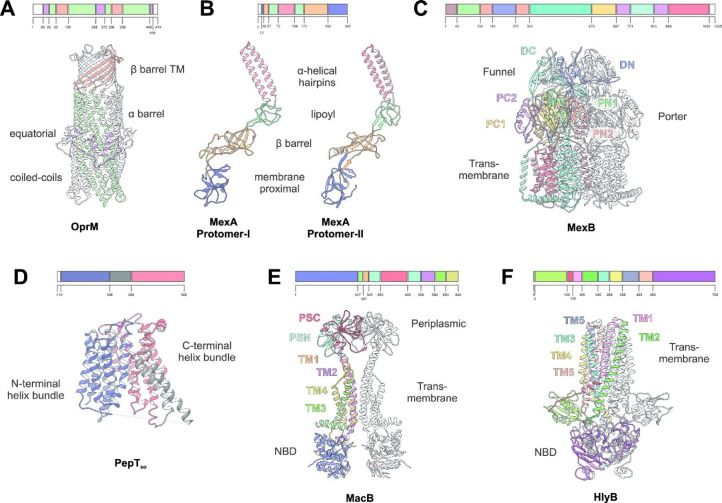
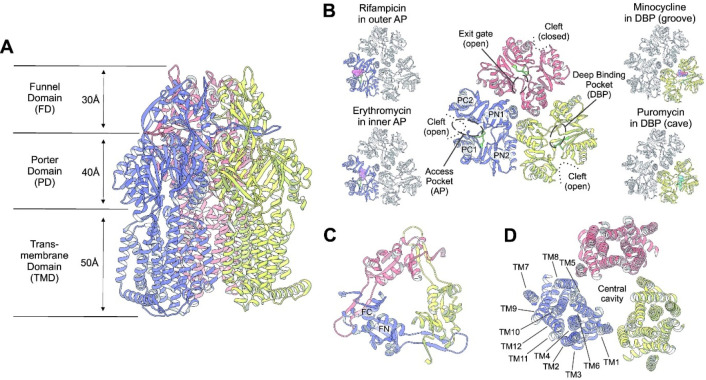
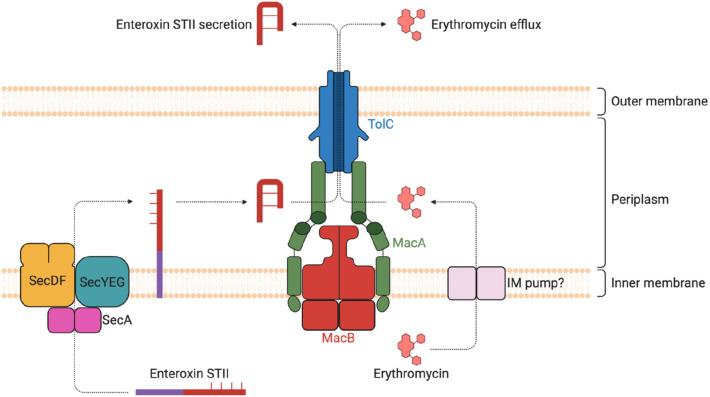
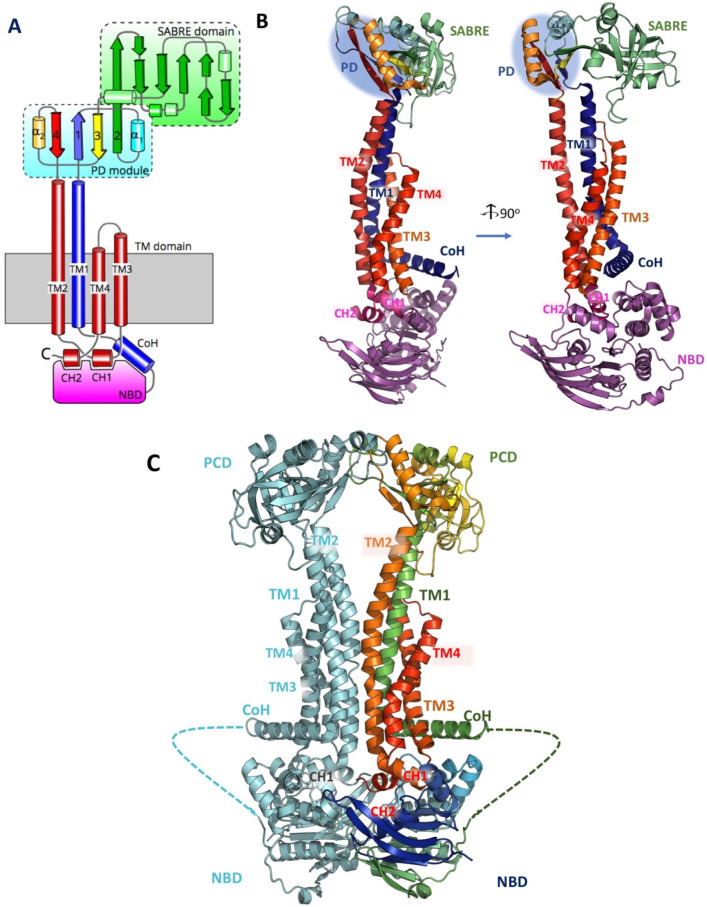
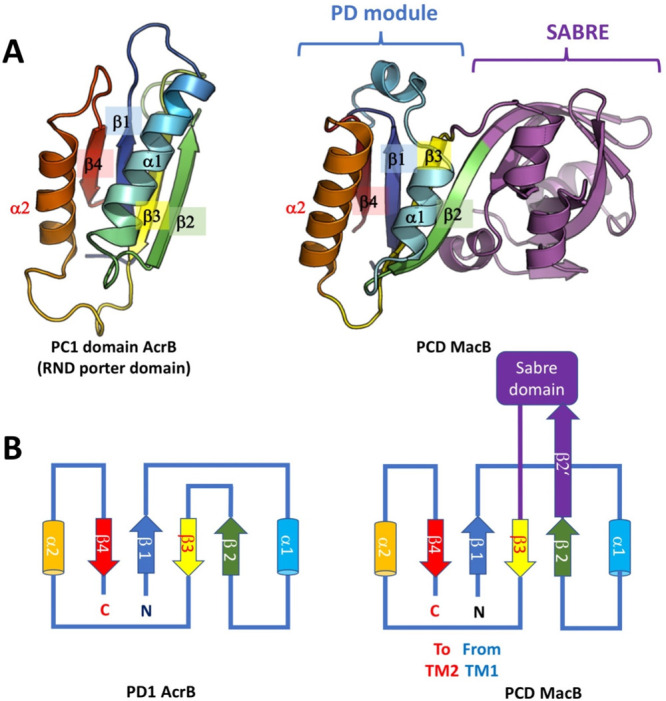
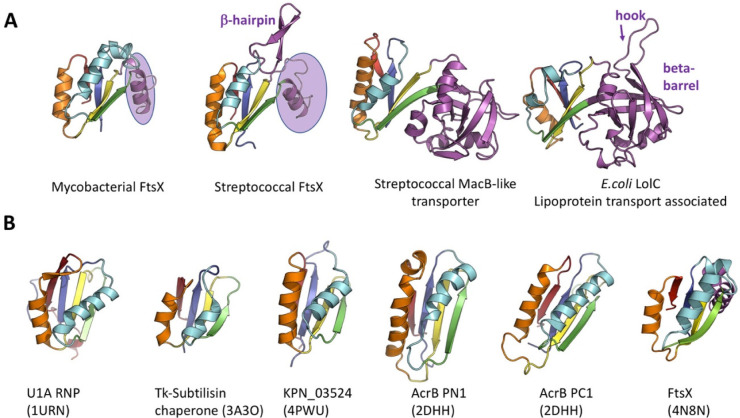
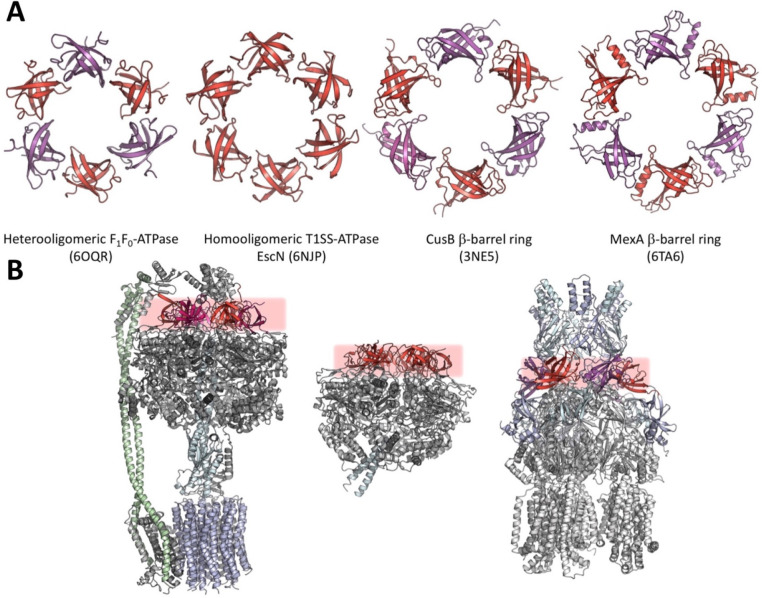
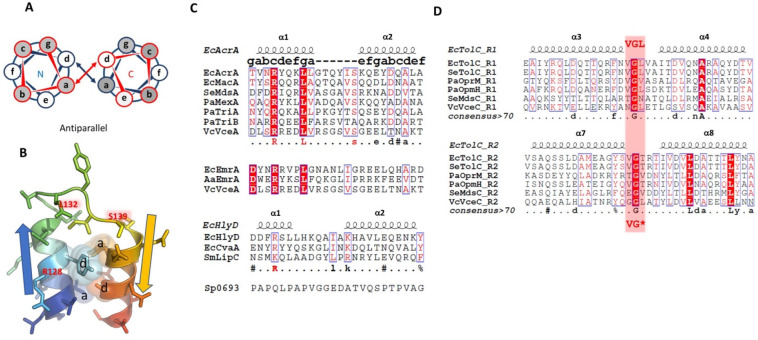
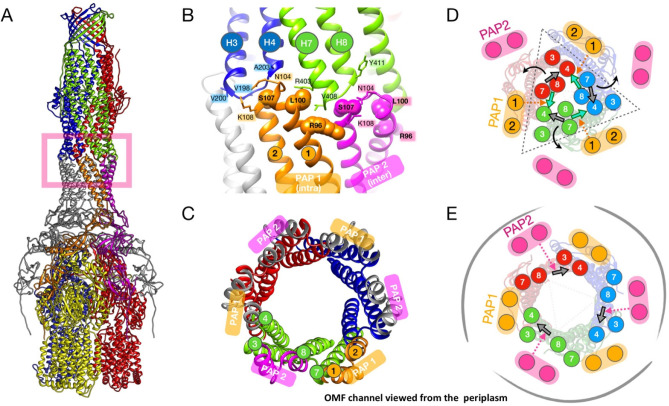
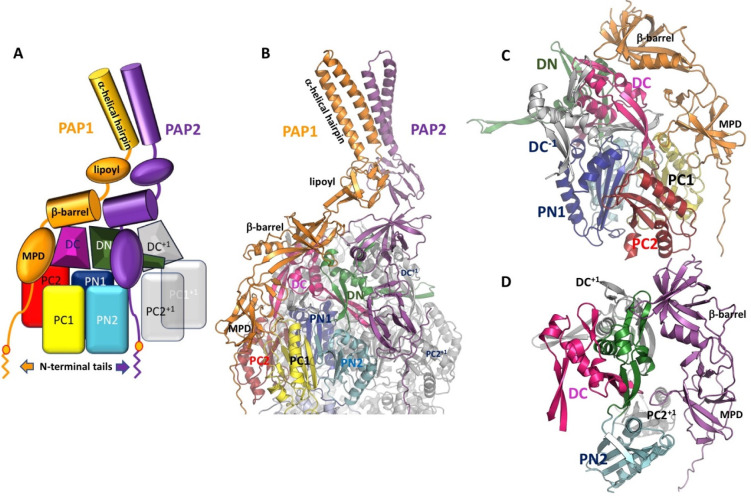
Tripartite efflux pumps and the related type 1 secretion systems (T1SSs) in Gram-negative organisms are diverse in function, energization, and structural organization. They form continuous conduits spanning both the inner and the outer membrane and are composed of three principal components-the energized inner membrane transporters (belonging to ABC, RND, and MFS families), the outer membrane factor channel-like proteins, and linking the two, the periplasmic adaptor proteins (PAPs), also known as the membrane fusion proteins (MFPs). In this review we summarize the recent advances in understanding of structural biology, function, and regulation of these systems, highlighting the previously undescribed role of PAPs in providing a common architectural scaffold across diverse families of transporters. Despite being built from a limited number of basic structural domains, these complexes present a staggering variety of architectures. While key insights have been derived from the RND transporter systems, a closer inspection of the operation and structural organization of different tripartite systems reveals unexpected analogies between them, including those formed around MFS- and ATP-driven transporters, suggesting that they operate around basic common principles. Based on that we are proposing a new integrated model of PAP-mediated communication within the conformational cycling of tripartite systems, which could be expanded to other types of assemblies.
Conflict of interest statement
The authors declare no competing financial interest.
Figures

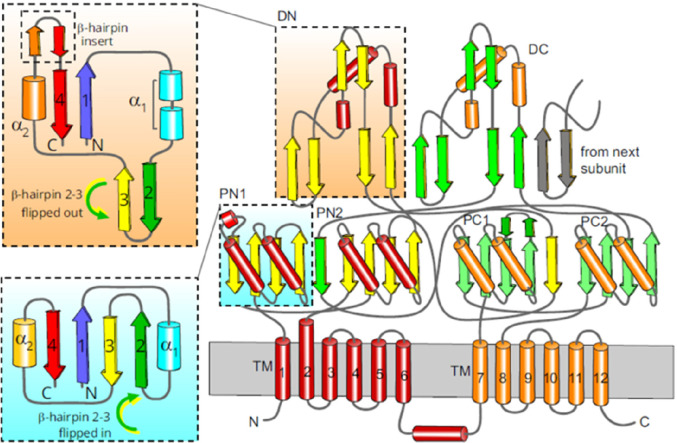
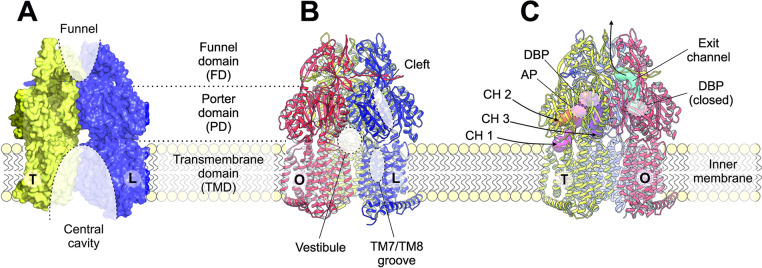
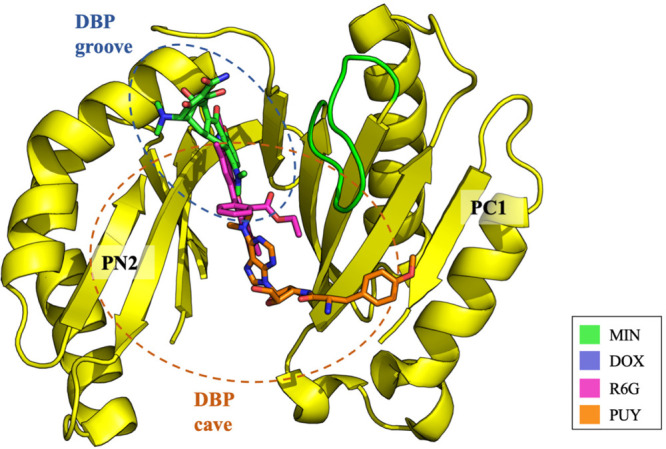
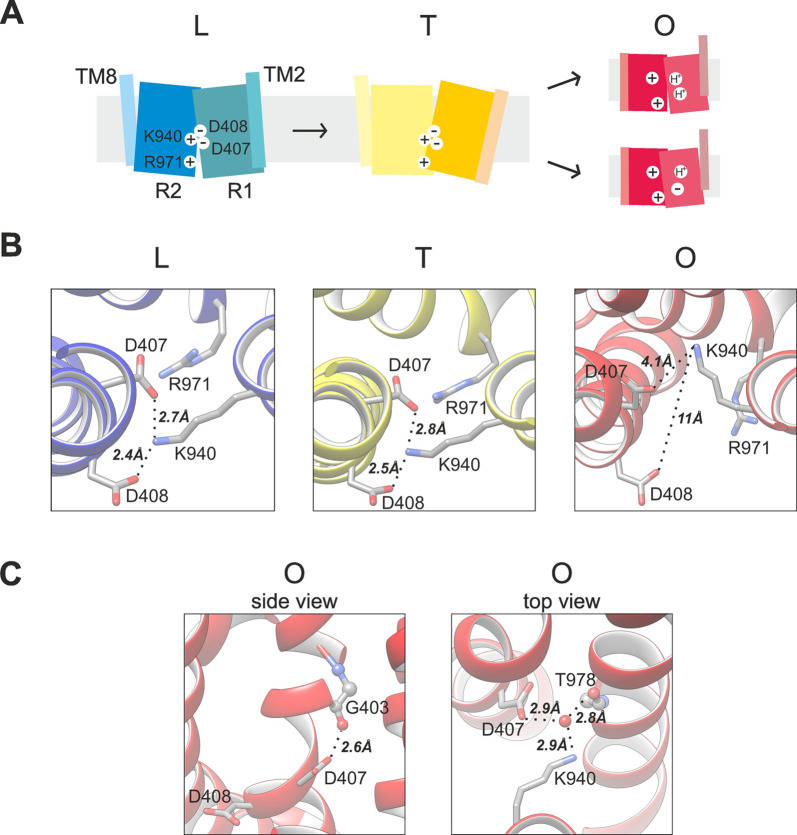
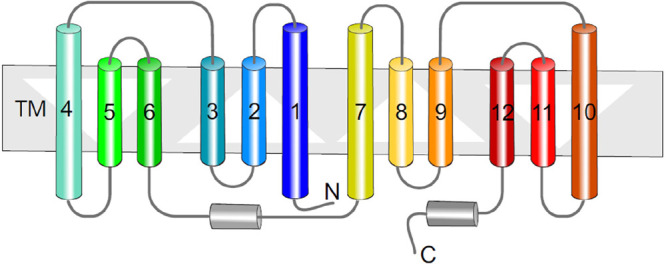
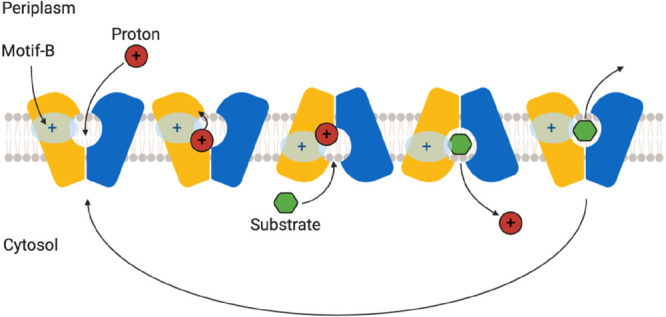
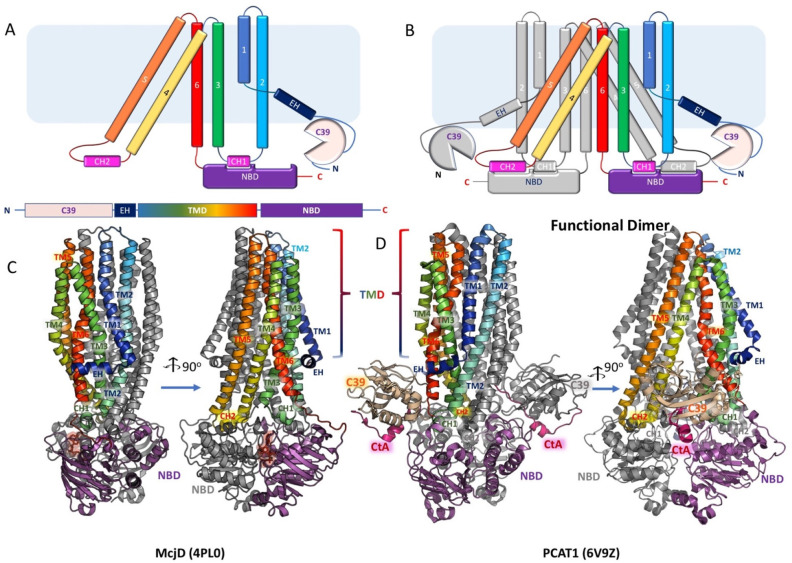
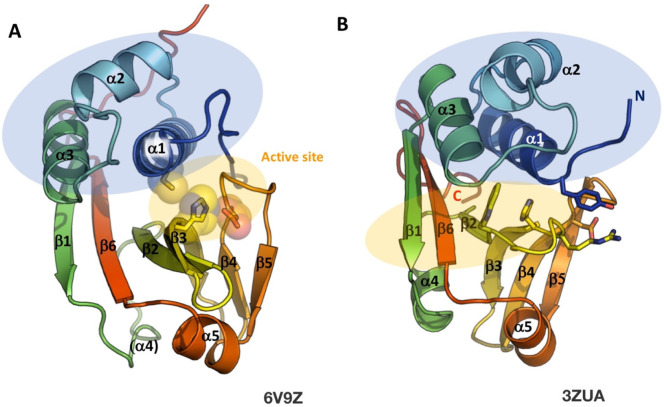
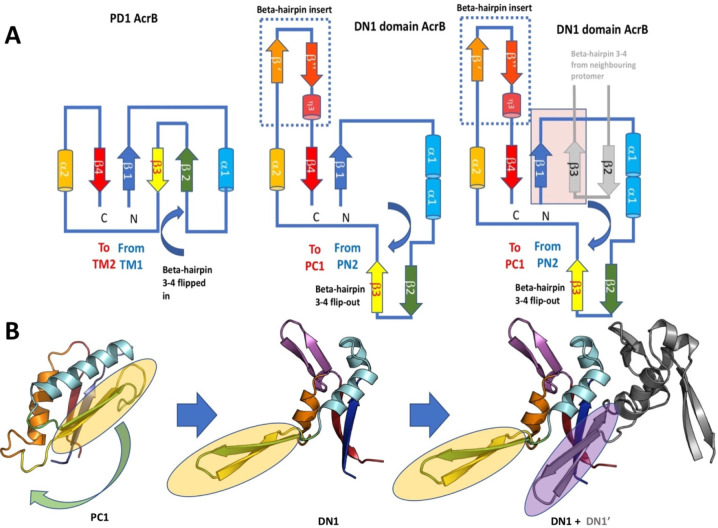
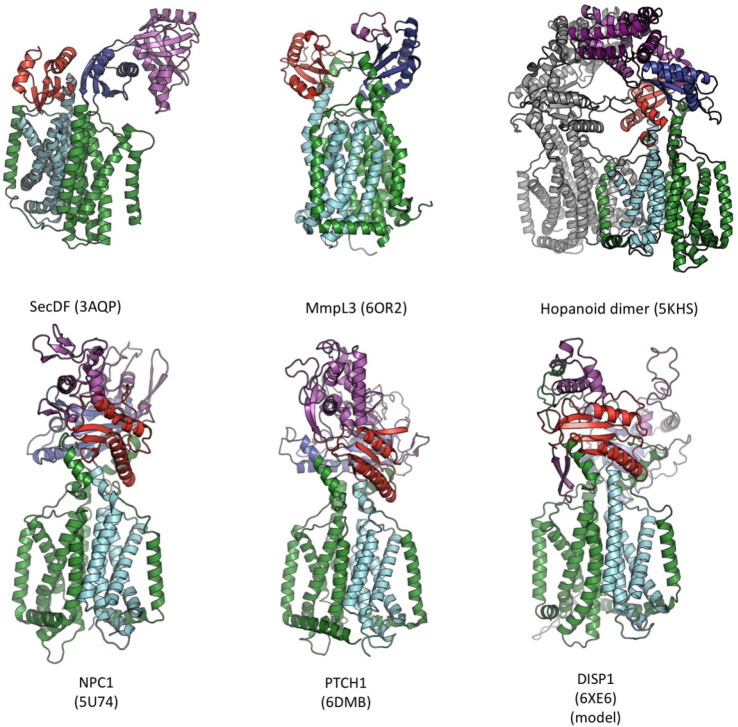
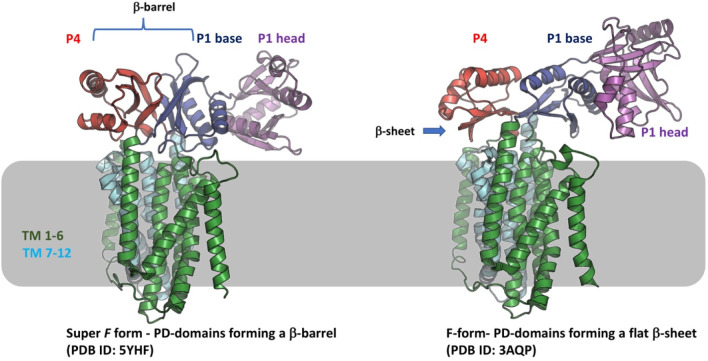
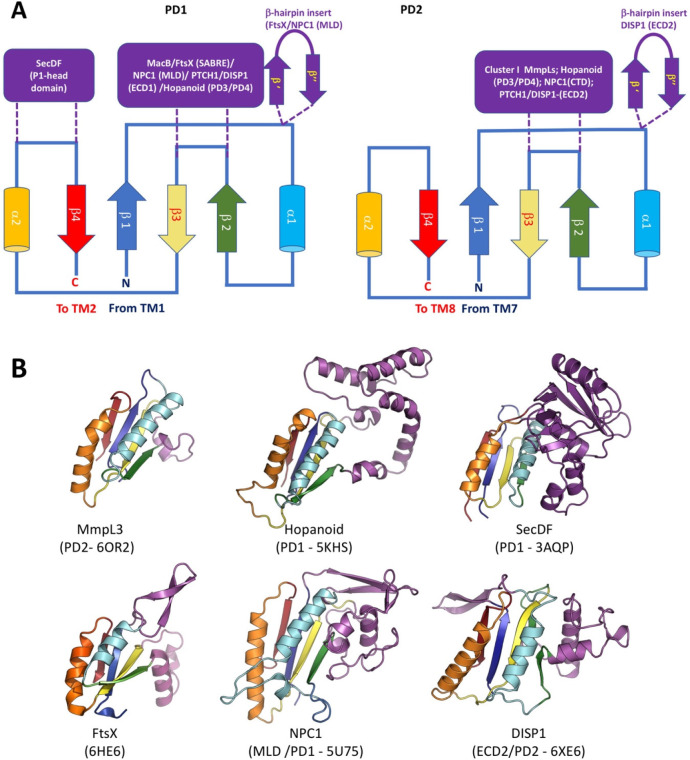
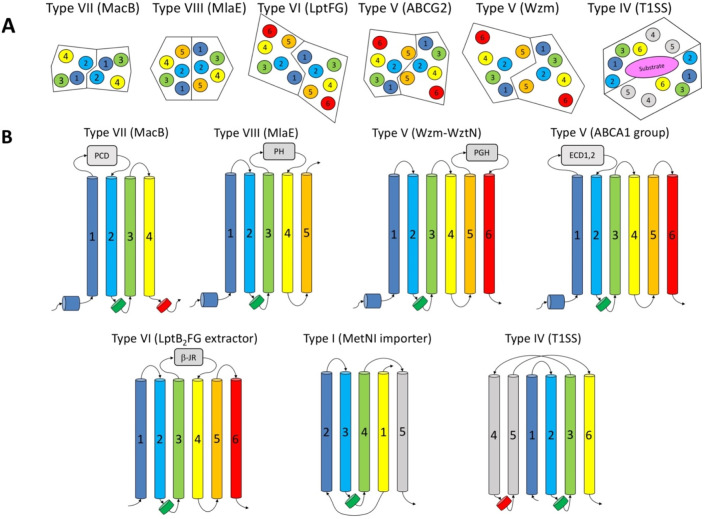
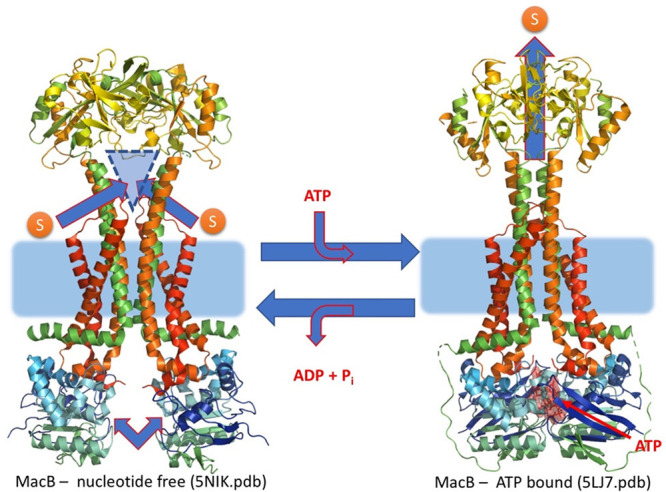
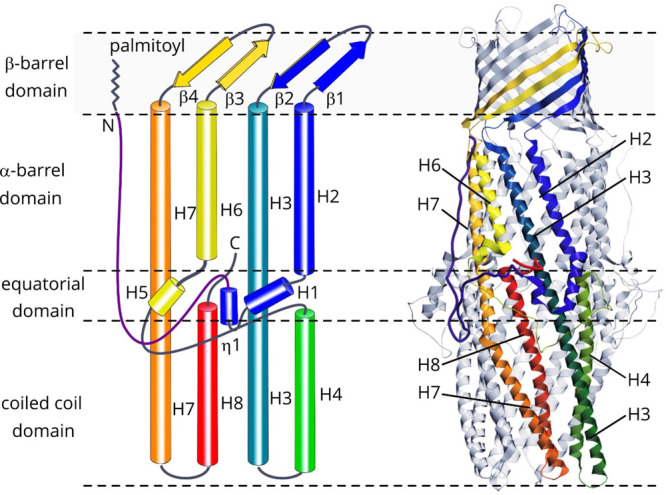
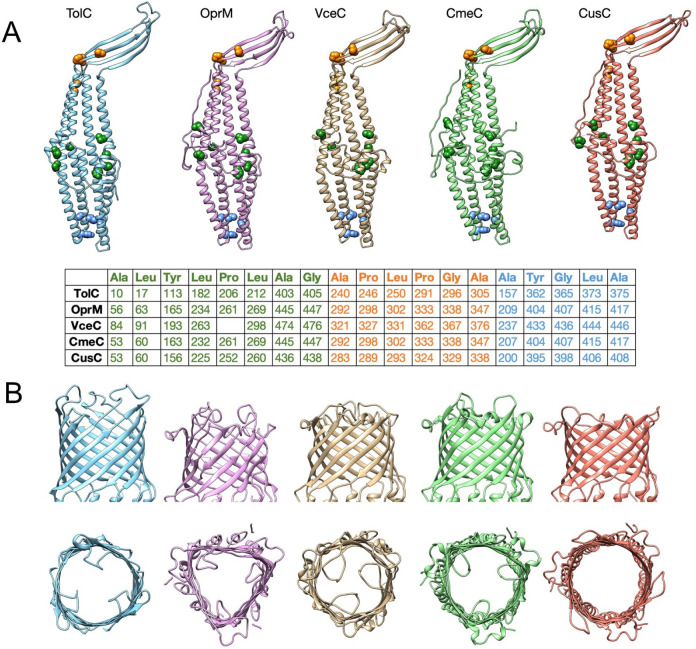
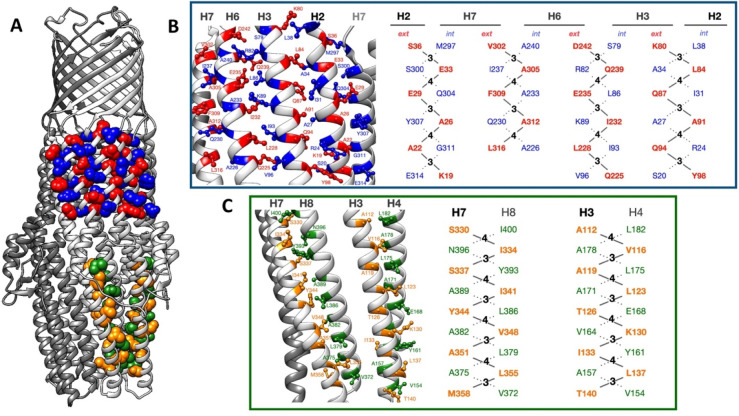
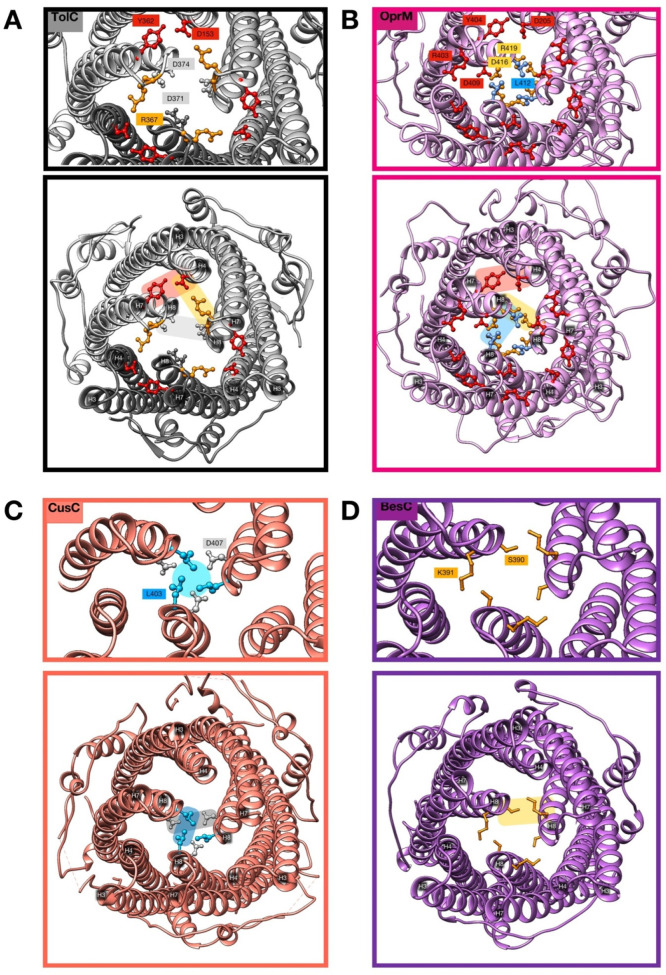
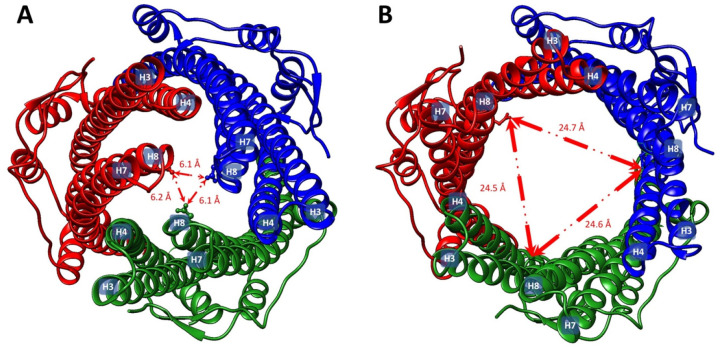
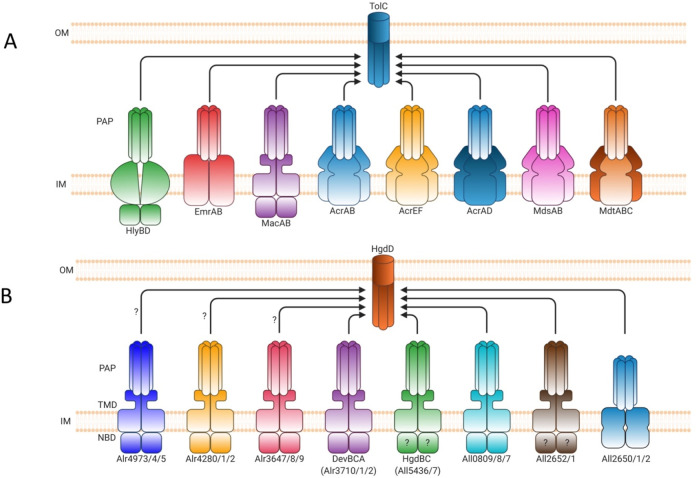

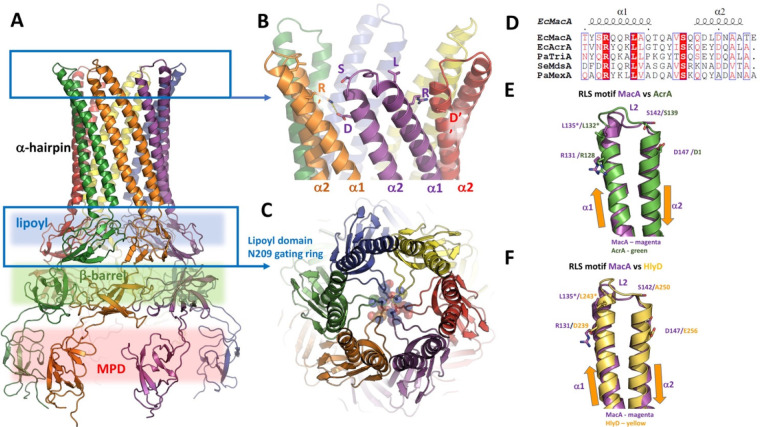
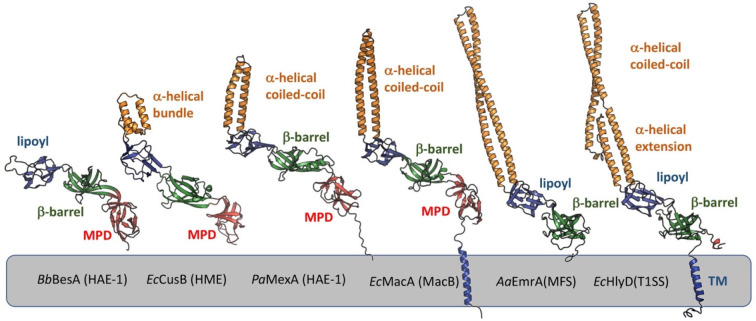
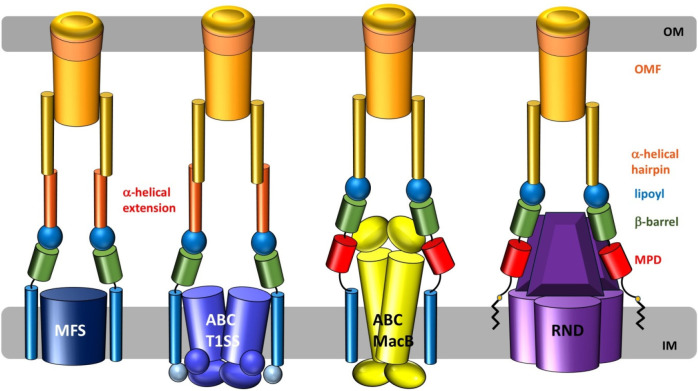

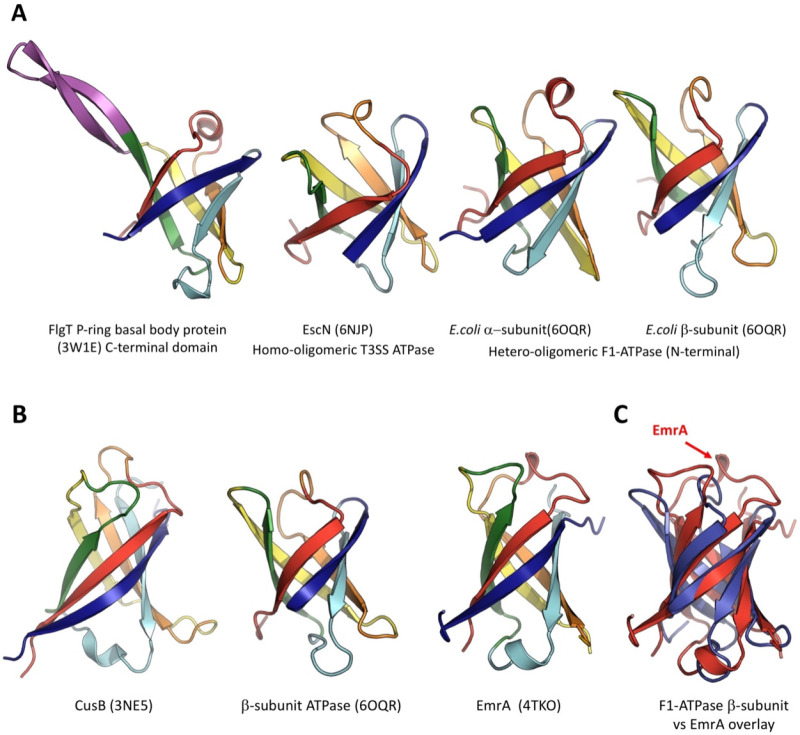
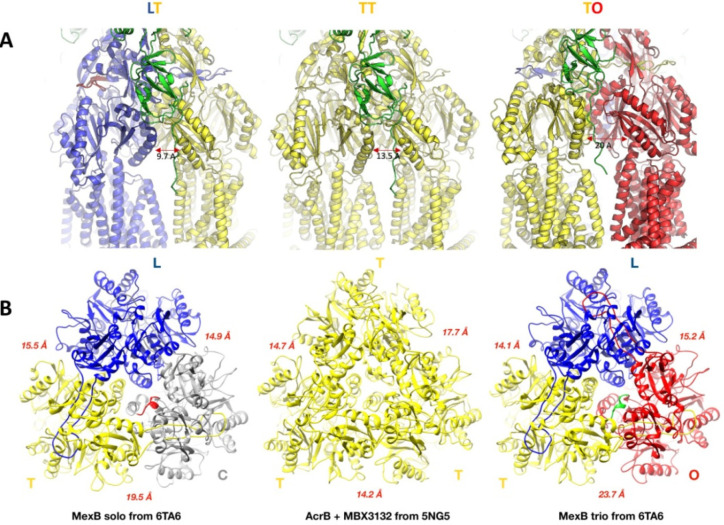
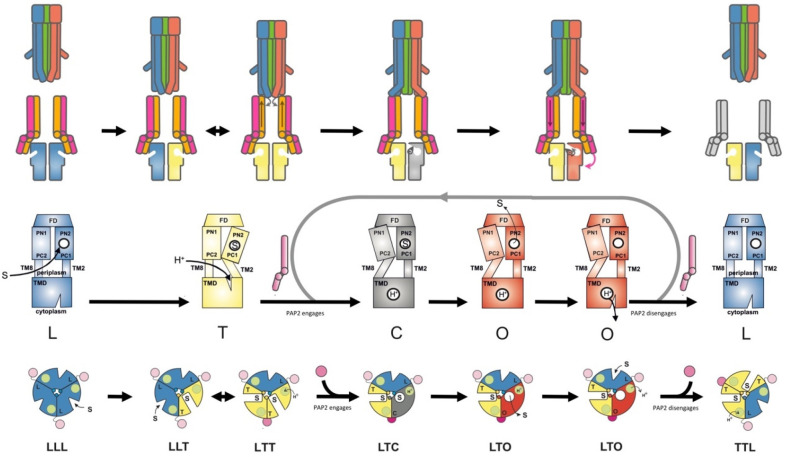
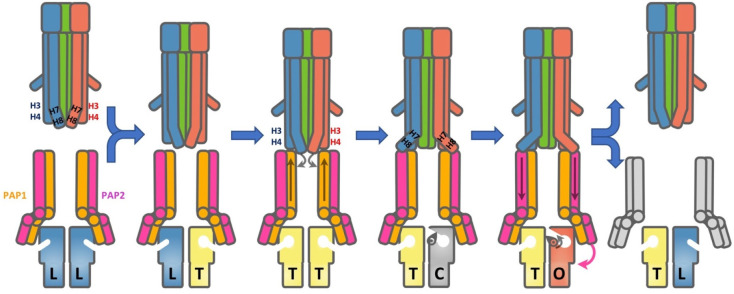
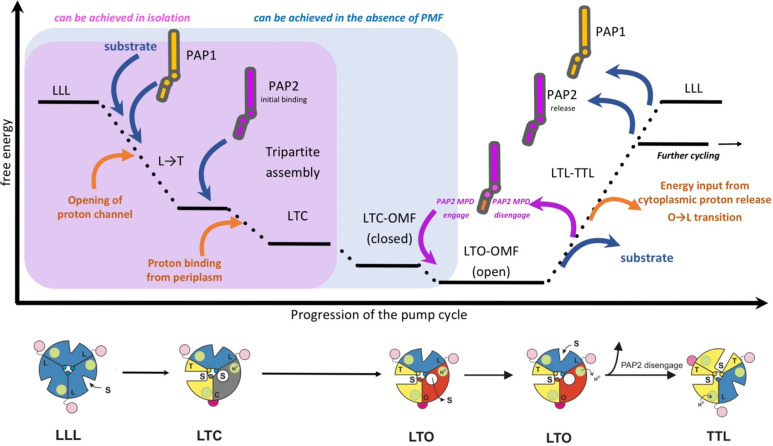
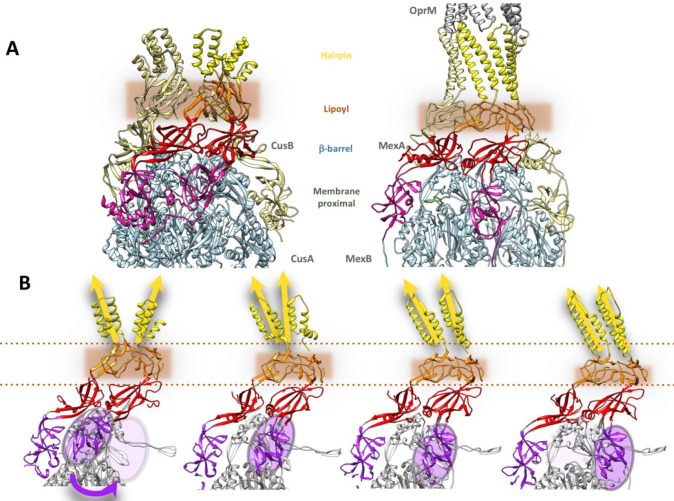
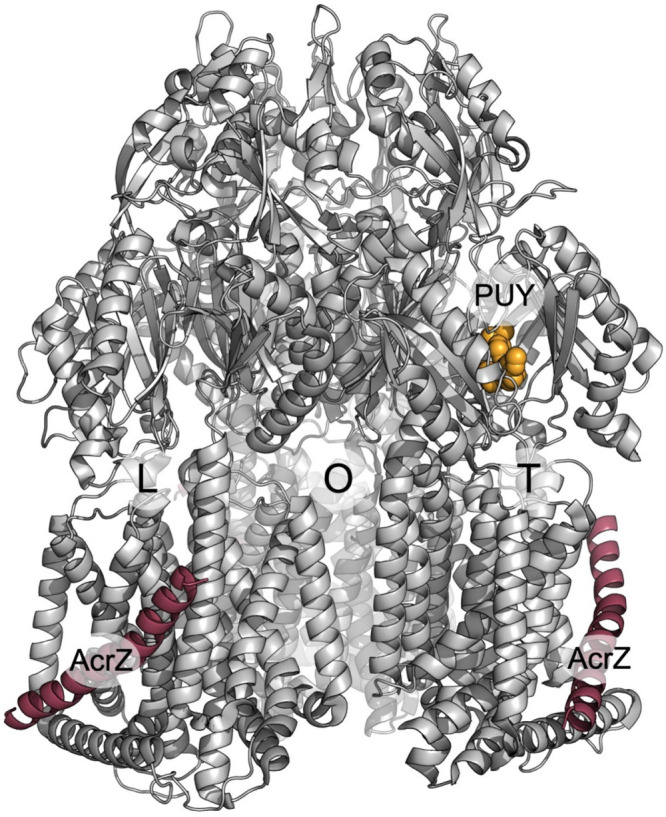
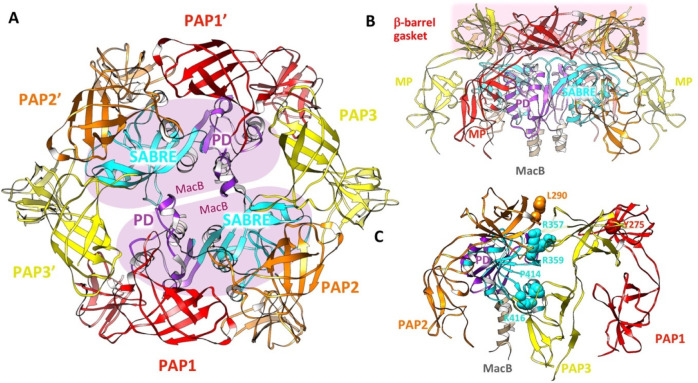
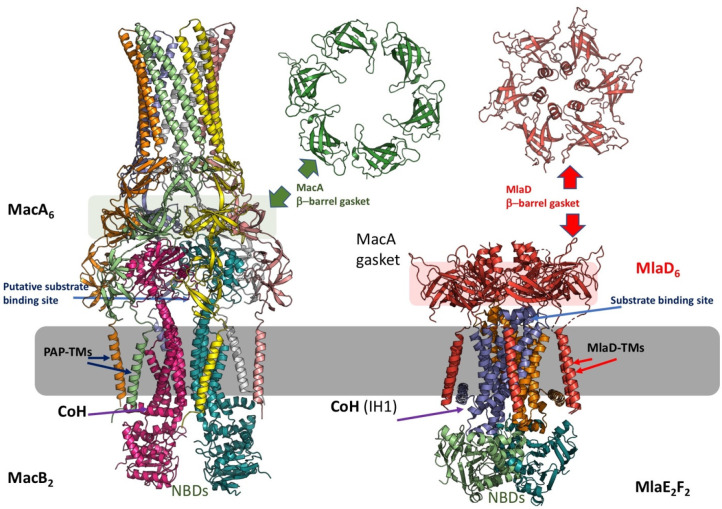
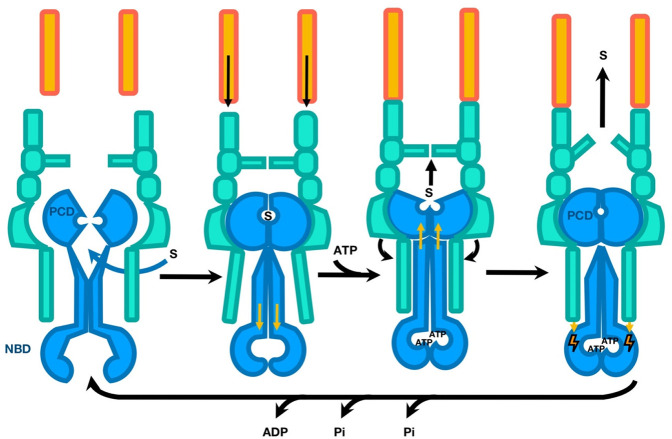
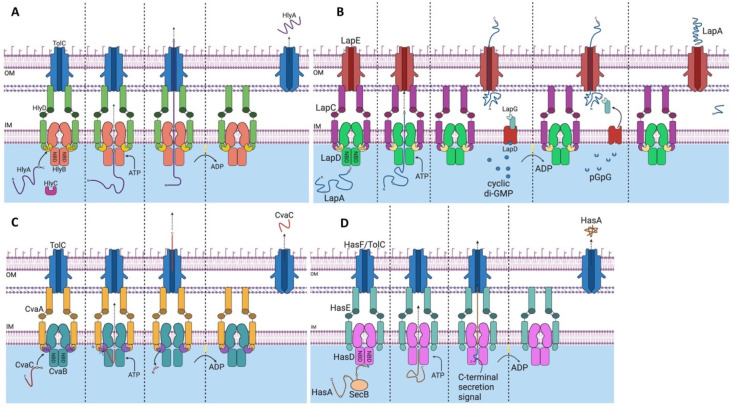
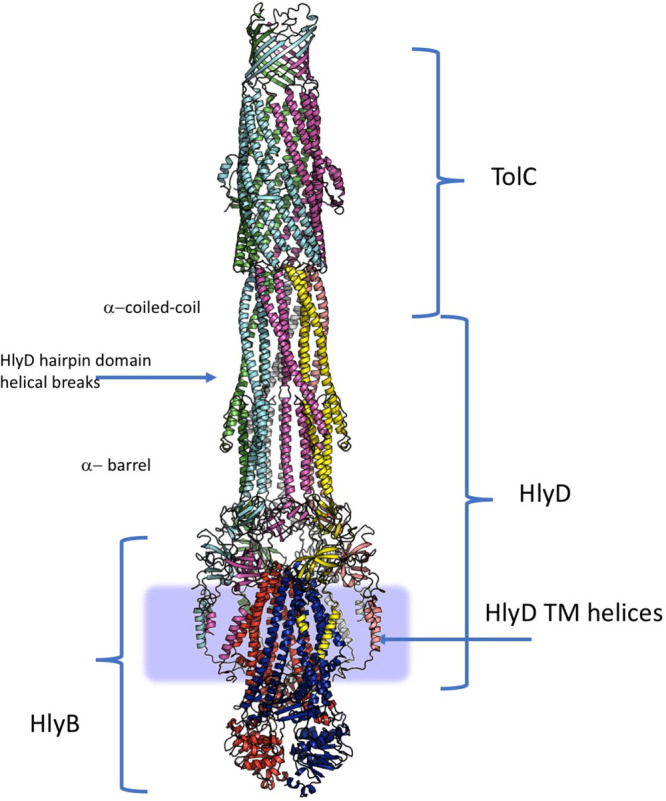
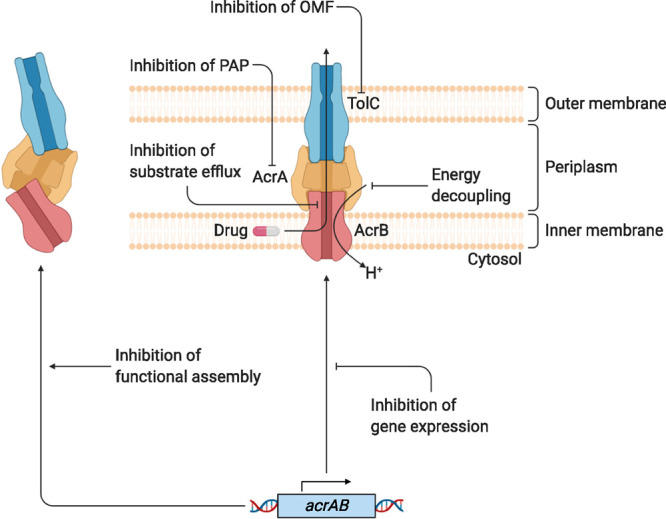
References
Publication types
MeSH terms
Substances
Grants and funding
- P41 GM103311/GM/NIGMS NIH HHS/United States
- BB/N002776/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/M02623X/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/M01116X/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
